Identification des bases génétiques derrière la
différence de réponse à un traitement chez un modèle
pour une maladie humaine
Mémoire
Véronique Hamel
Maîtrise en biologie - avec mémoire
Maître ès sciences (M. Sc.)
Identification des bases génétiques derrière la
différence de réponse à un traitement chez un
modèle pour une maladie humaine
Mémoire
Véronique Hamel
Sous la direction de :
Christian Landry, directeur de recherche
Marie Filteau, codirectrice de recherche
Résumé
En fonction du contexte génétique d’un individu et de son environnement, les mutations responsables de certaines maladies génétiques peuvent avoir des effets différents. Ces effets créent donc un besoin pour des traitements médicaux personnalisés. Ces traitements, pour être développés, demandent une meilleure compréhension de l’implication du contexte génétique au niveau moléculaire, incluant le mode d’action des gènes modificateurs. Pour mieux comprendre leur mode d’action, dans le cadre de mon projet, j’utilise un modèle du Syndrome de Wiskott-Aldrich chez la levure Saccharomyces cerevisiae impliquant le gène
LAS17. Grâce à des expériences d’évolution expérimentale, des gènes ciblés par des
mutations et, par la suite, une molécule ont été identifiés permettant de corriger le phénotype malade. Dans certains cas, cette correction est spécifique au contexte génétique et la majorité des gènes ciblés codent pour des partenaires d’interaction physique de Las17p. Une exception est la sous-unité Cnb1p de la calcineurine, la protéine ciblée par la cyclosporine A, qui ne fait pas partie de ces partenaires. La réponse à cette molécule est pourtant spécifique à un contexte génétique. J’ai utilisé différentes approches, dont des analyses QTL, pour identifier les gènes modificateurs sous-jacents à cette spécificité. Plusieurs locus ont été identifiés et la plupart incluaient des gènes au niveau du réseau d’interaction protéique ou génétique de Las17p et des sous-unités de la calcineurine. Le gène candidat principal, END3, un interactant physique de Las17p, semble présenter des effets de dosage au niveau protéique. Les liens avec la littérature suggèrent une importance dans la balance protéique de réseaux d’interaction à la fois du gène causant la maladie et celui ciblé par le traitement. L’ensemble des résultats du modèle soutiennent l’importance de tenir compte du contexte génétique pour élaborer de nouveaux traitements et suggèrent que les partenaires d’interactions devraient être les principales cibles de ces interventions.
Abstract
Depending on the genetic background of an individual and their environment, the mutations responsible for some genetic diseases may have different effects. These effects create a need for personalized medical treatments. To be developed, these treatments require a better understanding of the implication of the genetic background at the molecular level, including the mode of action of modifier genes. To better understand their mode of action, as part of my project, I used a model of the Wiskott-Aldrich Syndrome in the yeast Saccharomyces
cerevisiae involving the LAS17 gene. Through experimental evolution experiments, several
genes targeted by mutations and, subsequently, a molecule have been identified to rescue the diseased phenotype in yeast. In some cases, this phenotypic rescue was specific to the genetic context and the majority of the targeted genes coded for Las17p physical interaction partners. An exception was the Cnb1p subunit of calcineurin, the protein targeted by cyclosporin A, which was not one of these partners. The response to this molecule was nevertheless specific to a genetic background. I used different approaches, including QTL analysis, to identify modifier genes underlying this specificity. Several loci were identified and most of them included genes in the protein or genetic interaction networks of Las17p and of calcineurin subunits. The main candidate gene, END3, a physical interactant of Las17p, appeared to exhibit protein-level effects. The literature suggests an importance in the protein balance of the interaction networks of both the gene causing the disease and the one targeted by the treatment. The overall model results support the importance of considering the genetic background for developing new treatments and suggest that interaction partners should be primary targets for these interventions.
Table des matières
Résumé ... iii
Abstract ... iv
Table des matières ... v
Liste des figures ... vii
Liste des tableaux ... ix
Liste des abréviations et des sigles ... x
Remerciements ... xiii
Chapitre 1 - Introduction ... 1
1.1 Techniques pour déterminer les gènes impliqués derrière les maladies génétiques et diagnostiquer ces maladies ... 1
1.2 La médecine personnalisée ... 3
1.3 Les gènes modificateurs ... 3
1.4 Problématique : une seule souche de cellules, ou des populations consanguines ... 5
1.5 Les organismes modèles pour comprendre l’effet des gènes modificateurs ... 5
1.6 La levure comme organisme modèle ... 6
1.7 Un modèle de levure pour une maladie humaine : le Syndrome de Wiskott-Aldrich .. 7
1.8 Objectifs et hypothèses ... 12
Chapitre 2 - Matériel et méthodes ... 15
2.1 Milieux de culture ... 15
2.2 Souches ... 15
2.3 Immunobuvardage de la protéine Las17-41p ... 26
2.4 Tests de croissance en milieu liquide... 28
2.5 Croisement pour l’obtention de la souche BYxRM hétérozygote ... 31
2.6 Sporulation et dissection des souches ... 33
2.7 Test de croissance en milieu solide ... 33
2.7.1 Lavage avec une solution d’EDTA 30 mM ... 33
2.7.2 Patron de ségrégation et sélection des spores du croisement BYxRM hétérozygote ... 34
2.8 Préparation des pools de séquençage ... 35
2.9 Préparation de l’ADN et séquençage ... 36
2.10 Analyses des ségrégants en pool et gènes candidats ... 37
2.11 Analyse d’enrichissement des gènes candidats... 39
2.12 Réseaux d’interactions physiques et génétiques ... 39
2.13 Délétion des gènes candidats non-essentiels... 40
2.14 Test hémizygote réciproque ... 40
2.15 Échange d’allèle avec CRISPR-Cas9 ... 42
2.16 Validation des gènes candidats avec des plasmides ... 43
2.17 Vérification de l’effet des plasmides multicopies et centromériques sur la réponse à la CsA... 43
Chapitre 3 - Résultats ... 44
3.1 La CsA n’a pas d’impact direct sur la quantité de protéine las17-41p dans la cellule 44 3.2 La concentration optimale de CsA en milieu liquide : 10 µg/ml ... 46
3.3 Une combinaison des allèles de BY et de RM qui avantage la réponse à la CsA à 37°C ... 49
3.4 Les chromosomes XIII et XIV derrière la différence de réponse à la CsA ... 52
3.5 La perte d’une copie permet la modification de la réponse à la CsA ... 62
3.6 L’allèle END3 n’est pas directement responsable de la différence de réponse à la CsA ... 68
3.7 L’ajout d’une copie du gène END3 induit une réponse plus grande à la CsA... 71
3.8 Les plasmides centromériques permettent d’induire une réponse à la CsA chez RM 74 Chapitre 4 - Discussion... 79
4.1 L’influence du traitement n’est pas directe ... 79
4.2 Une réponse qui semble influencée par plusieurs gènes modificateurs ... 80
4.3 END3 comme gène modificateur par une différence d’abondance ... 82
4.4 MKT1 possiblement un gène modificateur ... 86
4.5 Une influence externe par des plasmides centromériques ... 87
4.6 D’autres traitements et contextes génétiques pour valider le modèle ... 89
4.7 L’importance du contexte génétique ... 91
Chapitre 5 - Conclusion ... 93
Bibliographie ... 95
Annexe I - Liste des ségrégants pour chacun des deux pools ... 106
Liste des figures
Figure 1.1 Alignement des séquences de WASP et ces homologues. ... 9 Figure 1.2 Phénotype de thermosensibilité de l’allèle las17-41 à 37ºC. ... 9 Figure 1.3 Correction du phénotype dû à la perte de fonction de las17-41 à 37ºC par l’inhibition de la calcineurine par la cyclosporine A (CsA) à une concentration de 10 µg/ml. ... 12
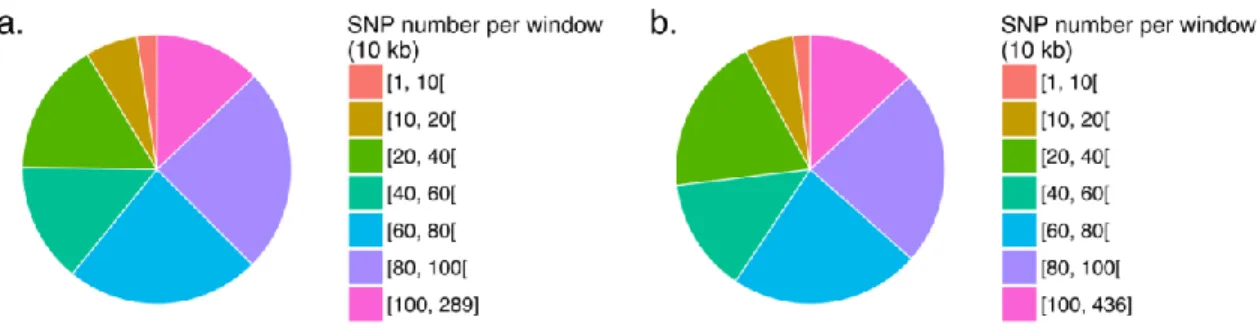
Figure 2.1 Méthode utilisée pour déterminer les bases génétiques de la variation de réponse à la CsA à 37ºC. ... 32 Figure 3.1 La cyclosporine A n’a pas d’impact direct sur la quantité de las17-41p produite aux températures de 25°C et de 37°C. ... 46 Figure 3.2 La concentration optimale de CsA pour la réponse des souches BY n’est pas différente de 10 µg/ml. ... 48 Figure 3.3 L’hybride BYxRM TS est avantagé dans sa réponse à la CsA face à celle de son parent BYxBY TS. ... 50 Figure 3.4 Plus de la moitié des ségrégants présentent une réponse à la CsA. ... 51 Figure 3.5 La majorité des fenêtres de 10 kb compte entre 10 et 100 SNPs. ... 54 Figure 3.6 Les chromosomes XIII et XIV représentent les principales régions responsables de la différence de réponse entre BY et RM. ... 55 Figure 3.7 Réseaux d’interactions physiques et génétiques entre les gènes candidats principaux, et LAS17 et les gènes codant pour les sous-unités de la calcineurine (CNA1,
CMP2 et CNB1). ... 62
Figure 3.8 La délétion des gènes APP1, MKT1 et NST1 n’affectent pas la réponse à la CsA à 37°C. ... 64 Figure 3.9 La perte d’une copie du gène END3 chez l’hybride et du gène MKT1 chez le diploïde RM modifie la réponse à la CsA. ... 67 Figure 3.10 Le changement d’allèle du gène END3 n’induit pas de changement au niveau de la réponse à la CsA. ... 71 Figure 3.11 L’ajout d’une copie de l’un ou l’autre des allèles de END3 induit une réponse plus grande que le plasmide pRS315 seul. ... 73 Figure 3.12 Réponse des souches BY et RM transformées avec des plasmides centromériques (pRS315, pRS316 et p5586) et des plasmides multi-copies (pRS425 et pRS426) en absence ou en présence de CsA (0 ou 10 µg/ml). ... 76 Figure 3.13 Certains des ségrégants composant les pools de réponse à la CsA et de non-réponse à la CsA ont conservé le plasmide pRS316, et cette conservation influence la fréquence allélique des locus responsables de la différence de réponse à la CsA... 78
Figure 4.1 Les gènes MKT1 et END3 sont adjacents l’un de l’autre, mais l’un est sur le brin sens et l’autre sur le brin anti-sens... 82 Figure 4.2 Plusieurs liens peuvent être faits avec des protéines impliquées dans la réponse au stress, l’organisation du cytosquelette d’actine et l’endocytose pour expliquer la réponse à la CsA et la différence d’abondance protéique de End3p. ... 86
Liste des tableaux
Tableau 2.1 Souches de S. cerevisiae utilisées et construites ... 16 Tableau 2.2 Plasmides utilisés et construits ... 20 Tableau 2.3 Amorces utilisées ... 21 Tableau 3.1 Position des locus déterminés comme étant potentiellement responsables de la différence de réponse à la CsA entre BY et RM sur les chromosomes du génome de BY et sur les contigs du génome de RM ... 56 Tableau 3.2 Gènes candidats ayant des interactions physiques ou génétiques avec Las17p ou les sous-unités de la calcineurine... 58 Tableau I. Liste des ségrégants utilisés pour chacun des deux pools suite à la sporulation de la souche BYxRM LAS17/las17-41-KanMX pRS316 ... 106 Tableau II.I Résultats du test d’enrichissement pour les termes GO sur YeastMine pour les processus biologiques pour les 122 gènes provenant de BY ... 109 Tableau II.II Résultats du test d’enrichissement pour les termes GO sur YeastMine pour les composants cellulaires pour les 122 gènes provenant de BY ... 115 Tableau II.III Résultats du test d’enrichissement pour les termes GO sur YeastMine pour les fonctions moléculaires pour les 122 gènes provenant de BY ... 124 Tableau II.IV Résultats du test d’enrichissement pour les termes GO sur YeastMine pour les processus biologiques pour les 32 gènes provenant de RM ... 125 Tableau II.V Résultats du test d’enrichissement pour les termes GO sur YeastMine pour les composants cellulaires pour les 32 gènes provenant de RM ... 135 Tableau II.VI Résultats du test d’enrichissement pour les termes GO sur YeastMine pour les fonctions moléculaires pour les 32 gènes provenant de RM ... 135
Liste des abréviations et des sigles
% : Pourcentage
°C : Degré Celsius
5-FOA : Acide 5-fluoroorotique
ANOVA : Analyse de la variance
ARS : Séquence de réplication autonome
BY : Contexte génétique de la souche BY4741 ou BY4742
BYxRM hétérozygote : BYxRM LAS17/las17-41-KanMX pRS316
CC : The Collaborative Cross
Cnap : Sous-unité calcineurine A
CsA : Cyclosporine A
CypA : Cyclophiline A
DGRP : The Drosophila Genetics Reference Panel
DO595/ml : Densité optique à 595 nm par ml
DMSO : Diméthylsulfoxyde
DTT : Dithiothréitol
EDTA : Éthylène Diamine Tétra-Acétique
FDR : False Discovery Rate
g : Gramme
G418 : Généticine
GO : Terme de Gene Ontology
GFP : Green Fluorescent Protein
H : Heure
Hyg : Hygromycine B
Kan : Kanamycine, ou marqueur de résistance
l : Litre
log2 ou log10 : Échelle logarithmique en base 2 ou 10
m : Mètre
M : Concentration molaire (mole par litre)
MATa : Type sexuel a MAT𝛼 : Type sexuel alpha min : Minute
Nat : Nourseothricine
NRCsA : Pool ne répondant pas à la CsA
PCA : Protein-fragment Complementation Assay
pb : Paire de bases
PCR : Réaction en chaîne par polymérase
PI4,5P2 : Phosphatidylinositol-4,5-biphosphate
QTL : Locus de caractères quantitatifs
RCsA : Pool répondant à la CsA
RM : Contexte génétique de la souche RM11
RPM : Rotation par minute
s : Seconde
Sc : Milieu de culture synthétique complet
SDS : Dodécylsulfate de sodium
SNP : Polymorphisme d’un seul nucléotide
Test-G : Test en log de rapport de vraisemblance
TP, ou RT : Température de la pièce, « Room Temperature », environ 22°C
TS : Thermosensible, las17-41 ou las17-41/las17-41
V : Volt
WAS : Syndrome de Wiskott-Aldrich
WT : Type sauvage, « Wild Type »
Remerciements
C’est avec un énorme plaisir que je vous présente aujourd’hui le travail que j’ai effectué au cours des deux dernières années et un peu plus pour ma maîtrise. À travers ces années, j’ai fait face à plusieurs embûches autant professionnelles que personnelles, dont certaines qui ont été pour moi des plus éprouvantes. Malgré tout, les jours, les heures, les minutes et les secondes qui ont été consacrés à mon projet de maîtrise ont été des plus enrichissants, et font partie de mes plus belles années. J’ai autant appris sur moi-même que sur les autres. Certains diront que ce n’est qu’une maîtrise, mais moi je dirai que ça a été plus. J’ai rencontré des personnes extraordinaires qui ont cru en moi et qui m’ont poussé jour après jour à continuer et à me dépasser.
Aujourd’hui, j’aimerais donc prendre le temps de remercier par écrit cesdites personnes qui ont façonné pour toujours la personne que je suis devenue. La toute première personne que j’aimerais remercier est mon directeur de recherche Christian Landry. J’aimerais te remercier d’avoir cru en moi dès le jour un, celui où tu as pris le temps de me passer en entrevue. Depuis ce jour, tu m’as poussé à croire en moi et en ce que je fais. Merci de ton écoute et de nos rencontres durant lesquelles tu prenais le temps d’écouter mes idées ! Merci de m’avoir donné autant de possibilités que je ne pourrais toutes les énumérer ! Une de celles dont je suis la plus fière, le talk que j’ai donné à la GSA ! Merci ! La toute première personne avec qui j’ai travaillé dans le Landry Lab a été Marie Filteau. Au cours des années, tu as toujours été là pour moi, pour m’écouter, pour me conseiller et je t’en remercie. Aujourd’hui, tu as ton propre lab et tu es ma co-directrice sur ce merveilleux projet, quoique pas toujours merveilleux, hein Chris ! Juste merci, car sans toi, comme c’est la suite de notre projet, il n’aurait probablement pas eu lieu !
Une des personnes chères à mes yeux qui a été là dès ma première journée en m’accueillant dans le laboratoire, qui a toujours été de bons conseils et qui prend toujours le temps, encore aujourd’hui, de répondre à mes questions et de m’écouter parler de mon projet et de mes problèmes personnels, est Alex. J’aimerais donc le remercier chaleureusement pour toutes ces qualités qui font de lui un professionnel de recherche unique. Et non, je n’ai rien à vous cacher à toi et à Gil aujourd’hui ! Une autre personne qui a aussi toujours pris de son temps
pour me conseiller et m’écouter autant sur le point personnel que professionnel, mais qui n’a pas toujours été présente au laboratoire, à cause bien entendu de ces deux grossesses qui ont mis au monde deux beaux petits garçons, est Isabelle, notre professionnelle de recherche. Cette maman du lab, comme Alex aime bien l’appeler, a été là pour m’aider d’en bien des moments et je tiens également à la remercier chaleureusement.
Au cours de mon passage au laboratoire, plusieurs personnes ont passé : Guillaume Diss, Luca, Sam, Diana, Martha, Chloé, Mariam, JB, Hélène Vignaud, Francis, Mani, André-Ève, FO, Nabila, Éléonore …, toutes des personnes qui à différents niveaux ont été là pour moi et qui m’ont écoutée, avec qui j’ai parlé de tout et de rien, autant au Tea Break qu’en congrès ou à d’autres moments. Surtout, Andrée-Ève et Éléonore, merci ! Mais bon le point n’était pas là, la seule personne autre que nos professionnels de recherche à être encore là, est Gil ! Je voulais te dire merci ! Merci d’être toi, et d’avoir été là pour moi comme tu l’as fait durant ces dernières années ! Merci pour ton écoute, ta compréhension, ton humour ! Juste merci ! Merci également à Phil, pour ton écoute et les bons moments dans le lab, et pour réparer la cruche, on a été une équipe d’enfer ! Merci aussi à Caro, Carla, Simon, Yacine, Marie, Luca, Mariam, Mani, Nabila, Andrée-Ève et Éléonore (encore une fois !) qui ont tous à un moment ou à un autre partagé le bureau 3212 avec moi et ont dû m’écouter parler de divers sujets ! Merci également à Axelle, Mathieu, Chris, Souhir, Claudine, Clara et Anna qui ont su faire la différence quand j’en avais le plus besoin et qui ont été là pour me supporter à divers moments ! Merci également à François, Guillaume N., Matteo, Angel, Pauline et Ugo pour les bons moments ! Merci à Lou d’avoir pris le temps de répondre à mes questions en statistique et sur d’autres sujets, bien évidemment ! Un merci tout spécial à Hélène Martin ! Merci pour ton aide avec R et mes graphiques ! Merci pour toutes les choses que tu as faites pour moi et merci pour ton temps qui a été si précieux à mes yeux ! Merci pour les longs moments au pub à discuter, avec Lou et Gil, bien entendu !
J’aimerais également remercier Yves Bourbonnais et Louis Bernatchez qui ont été sur mon comité d’encadrement ! Merci de vos précieux conseils sur mon projet au tout début ! Merci à Yves qui prend encore le temps aujourd’hui de me demander des nouvelles sur son avancement !
Merci à tous les membres de l’IBIS et du département de biologie qui de près ou de loin ont su faire la différence !
Merci au CRSNG et au FRQNT d’avoir voulu me supporter financièrement pour ce projet !
Mes derniers remerciements, j’aimerais les désigner à ma famille proche. À ma mère tout particulièrement sans qui je ne serais probablement pas ici ! Merci pour tes encouragements, pour la manière que tu as de m’écouter même si tu ne comprends pas toujours ce dont je parle ! Merci d’avoir été là à tous les jours, pour partager mes bons moments et mes pleurs ! J’ai eu peur, très peur pour toi, mais tu es encore là ! Merci de croire en moi, en mes rêves ! Merci tout simplement d’être toi et d’être là ! Je t’aime plus gros que l’univers ! Merci à mes grands-parents ! Merci d’avoir été là pour votre écoute et vos conseils ! Merci ! Merci à mon frère et à mon père ! Merci pour les moments partagés et d’avoir été là, juste là ! Merci !
Chapitre 1 - Introduction
1.1 Techniques pour déterminer les gènes impliqués derrière les maladies
génétiques et diagnostiquer ces maladies
Les techniques pour mieux comprendre les bases génétiques derrière les maladies humaines et pour les diagnostiquer sont devenues de plus en plus puissantes. En effet, grâce au progrès du génie génétique, le séquençage de nouvelle génération permet de séquencer un génome entier en peu de temps pour obtenir un diagnostic (Koboldt et al., 2013). Le traitement de cette forme de diagnostic doit toutefois passer d’abord par des études expérimentales qui permettent de déterminer les gènes impliqués derrière ces maladies. L’avancement de la technologie a entre autres permis de passer des études de liaisons génétiques à des études d’association pangénomique (« Genome-Wide Association Study »; GWAS) puis à un séquençage complet du génome pour identifier les mutations ou les indels responsables des maladies génétiques (Koboldt et al., 2013).
Les études de liaisons génétiques se basent sur la découverte des gènes responsables de la maladie par l’étude de familles complètes en tenant en compte de la transmission des allèles à travers les générations. Elles fonctionnent donc bien pour des maladies impliquant un seul gène qui sont hautement pénétrantes dans la population (Koboldt et al., 2013). Un bon exemple est celui de la fibrose kystique, qui implique le gène CFTR, pour laquelle plusieurs études de liaisons génétiques à travers des familles ont montré un lien avec ce gène (Kerem
et al., 1989). Par contre, ce type d’approche ne permet pas d’identifier des variants dans la
population responsables de maladies beaucoup plus complexes au niveau génétique (Koboldt
et al., 2013).
L’approche GWAS a apporté avec elle une méthode puissante permettant de mieux discerner le signal des variants impliqués dans les maladies génétiques complexes. En effet, cette approche se basant sur l’association de variations génétiques à un phénotype donné par l’étude de plusieurs individus a permis d’associer plusieurs allèles à ces maladies génétiques
(Risch and Merikangas, 1996). Le GWAS a été utilisé, et est encore utilisé, dans plusieurs domaines d’étude incluant l’étude de structure de population (Pariyar et al., 2016), des traits anthropométriques (Berndt et al., 2013) et, évidemment, des maladies complexes, tel que la maladie de Crohn (Yamazaki et al., 2005). D’ailleurs, un catalogue des données suivant des études GWAS a été établi pour être en mesure de prédire le rôle de variants communs pour des maladies complexes (Hindorff et al., 2009). Selon Koboldt et al. (2013), le GWAS est probablement l’approche qui a eu le plus d’impact au niveau clinique par le biais de la pharmacogénomique. L’ajustement de la dose de warfarine, un anticoagulant, basé sur la variation génétique des gènes VKORC1, CYP2C9 et CYP4F2 est un exemple de son application (Takeuchi et al., 2009). Étant donné la nature de l’approche, contrairement à la méthode de liaison génétique, le GWAS a généralement donné lieu à l’identification de peu de variants à larges effets (Koboldt et al., 2013).
Avec le développement des approches de séquençage de nouvelle génération et la combinaison des diverses méthodes déjà établies, les possibilités sont de plus en plus grandes. En effet, le séquençage de génomes entiers permet non seulement l’identification de plusieurs variants communs qui peuvent avoir peu ou moyennement d’effets sur le phénotype (Modèle de maladie commune – variant commun) (Chakravarti, 1999; Lander, 1996; Lohmueller et
al., 2003), mais l’identification de variants plus rares qui peuvent avoir de forts effets
(Modèle de maladie commune – variant rare) (Cohen et al., 2006; Manolio et al., 2009; Zhu
et al., 2010). Ainsi, le séquençage de nouvelle génération permet de faire la détermination
d’un plus grand nombre de gènes responsables de certaines maladies génétiques en couvrant les différents types de variants. Cette connaissance permet ensuite de faire le diagnostic de maladies génétiques par la même approche chez les individus présentant les symptômes de la maladie, et même d’en faire le diagnostic prénatal (Koboldt et al., 2013).
1.2 La médecine personnalisée
Avec le diagnostic des maladies génétiques et la connaissance des gènes responsables de certaines de ces maladies, une nouvelle approche de la médecine fait maintenant son entrée : la médecine personnalisée. La médecine personnalisée est une forme de traitement du patient qui tient en compte que chaque individu est différent, à la fois au niveau génétique et au niveau de son environnement. Cette différence entre les individus implique que, pour une même maladie, chaque individu peut présenter des symptômes différents et que la réponse à un traitement donné de ces mêmes individus peut également être différente (Chow, 2016; Lu
et al., 2014). Plusieurs exemples de maladies sont retrouvés dans la littérature démontrant
que le contexte génétique des individus influence à la fois les symptômes et les traitements de la maladie, dont la fibrose kystique (Chow, 2016), le cancer (Ferguson et al., 2015), la lipodystrophie (Vantyghem et al., 2004) et l’hépatite C (Ge et al., 2009).
1.3 Les gènes modificateurs
L’une des composantes du contexte génétique qui est derrière la différence de symptômes pour une même maladie, ou encore la réponse à un même traitement est le gène modificateur. Les gènes modificateurs sont des gènes qui viennent influencer l’effet des mutations. Au niveau d’un phénotype délétère causant une maladie génétique, ils peuvent influencer notamment la gravité des symptômes chez les individus (Nadeau, 2001). Il y a donc une interaction entre la mutation causant la maladie et les gènes modificateurs qui composent le contexte génétique (Lehner, 2013). Les gènes modificateurs peuvent affecter le phénotype autant au niveau de son expression, de la pénétrance dans la population, de la modification de la dominance et de la pléiotropie (Nadeau, 2001). Par leur interaction avec la mutation, ces gènes peuvent autant avoir un effet négatif sur celle-ci, donc, par exemple, en augmentant les symptômes de la maladie, ou encore, avoir un effet positif, donc diminuer les symptômes, et dans certains cas, même les effacer (Kammenga, 2017).
L’une des maladies humaines pour laquelle plusieurs gènes modificateurs ont été identifiés pour expliquer les différences phénotypiques est la fibrose kystique (Gallati, 2014). Entre les
individus, pour cette maladie, il y a une grande variabilité autant au niveau de la sévérité de la maladie, des complications qui y sont associées et de la mortalité (Cutting, 2015). D’ailleurs, une étude comparant des jumeaux pour connaître l’implication de plusieurs composantes derrière des différences de mesures cliniques de la fonction pulmonaire a montré qu’environ 50% étaient attribuables au facteur génétique (Collaco et al., 2010). Ainsi, une étude menée par le North American Cystic Fibrosis Gene Modifier Consortium, qui a utilisé le GWAS et la liaison génétique sur un total de 3 467 patients, a réussi à identifier des locus associés à la sévérité de la maladie pulmonaire et dont les gènes dans ces régions ont un lien avec la fibrose kystique (Wright et al., 2011). Suite à cette étude, le gène EHF a été confirmé comme étant un gène modificateur altérant la capacité des cellules épithéliales à effectuer correctement le repliement et le trafic des mutants CFTR (Stanke et al., 2014). Un autre gène dans la même région chromosomique, le gène APIP, est suggéré comme empirant les symptômes de la fibrose kystique lorsque son expression est augmentée étant donné l’augmentation de l’inhibition de l’apoptose, l’une de ses fonctions (Wright et al., 2011). Ces deux gènes sont des exemples parmi plusieurs autres pour cette maladie (Cutting, 2015; Gallati, 2014).
Les gènes modificateurs peuvent également avoir des effets positifs, comme montré dans une étude récente de Chen et al. (2016) où des individus ont été retrouvés dans la population humaine comme présentant des variants responsables de certaines maladies génétiques, mais ne présentant aucun symptôme (Chen et al., 2016). Les maladies génétiques étudiées étaient des maladies infantiles mendéliennes connues comme étant hautement pénétrantes dans la population. Pourtant, ces individus résilients semblent présenter un contexte génétique leur permettant de ne pas développer les symptômes liés à la maladie. Il faut toutefois garder à l’esprit que pour des raisons de confidentialité dans les données utilisées pour cette étude, les cas n’ont pas pu être validés. Malgré tout, l’étude de cas comme ceux présentés dans cette étude peut non seulement permettre de comprendre l’impact du contexte génétique dans la différence de symptômes pour une même maladie, mais également être l’avenue vers de nouveaux traitements en ciblant les gènes modificateurs.
1.4 Problématique : une seule souche de cellules, ou des populations
consanguines
Malgré cette connaissance de l’effet du contexte génétique et de ses gènes modificateurs, il y a tout de même un problème qui persiste dans les approches expérimentales utilisées. Pour étudier des maladies et leurs traitements, notamment à partir d’organismes modèles, la plupart des études utilisent une seule souche de cellules, ou encore des populations consanguines. La variabilité génétique retrouvée dans la population humaine n’y est donc pas bien représentée (Chow, 2016; Ferguson et al., 2015). Cette condition a pour effet que les études visant à trouver des traitements potentiels ciblant une voie qui affecte la maladie trouvent des traitements fonctionnels seulement pour une partie de la population. Une solution proposée pour arriver à contourner ce problème est d’étudier la différence entre les contextes génétiques et leurs effets tout en tenant compte de l’environnement des individus (Chow, 2016; Eichler et al., 2010).
1.5 Les organismes modèles pour comprendre l’effet des gènes
modificateurs
Pour arriver à mieux comprendre l’effet du contexte génétique et des gènes modificateurs, plusieurs études ont justement utilisé les organismes modèles. En plus d’avoir certains avantages permettant de tester directement les solutions trouvées, les organismes modèles permettent, dans une certaine mesure, d’avoir une vue simplifiée du modèle complexe qu’est l’humain. Avec les connaissances et les outils développés pour chacun de ces organismes, il est donc possible d’essayer d’investiguer davantage les processus moléculaires derrière les maladies génétiques complexes. Parmi les organismes modèles utilisés pour étudier ces maladies, on retrouve la souris (Mus musculus), le poisson zèbre (Danio rerio), la mouche à fruit (Drosophila melanogaster), le nématode (Caenorhabditis elegans) et la levure (Saccharomyces cerevisiae) (Lehner, 2013; Strynatka et al., 2018).
Dépendamment des outils développés, certains organismes modèles permettent d’avoir une bonne représentation de la variation génétique humaine. En effet, des collections d’individus
permettant cette représentation ont été élaborées telles que The Collaborative Cross (CC) chez la souris (Churchill et al., 2004; Morahan et al., 2008; Threadgill et al., 2002; Welsh et
al., 2012) et The Drosophila Genetics Reference Panel (DGRP) chez la mouche à fruit
(Huang et al., 2014; Mackay et al., 2012). Ainsi, l’utilisation de ces collections dans différentes études a permis d’étudier des cas de maladies humaines dans différents contextes génétiques (Chow et al., 2016; Ferguson et al., 2015; He et al., 2014; Park et al., 2014). Par exemple, pour la maladie de dégradation du rétinal, Retinitis pigmentosa, Chow et al. (2016) ont montré que les manifestations phénotypiques de la maladie chez la mouche à fruit sont effectivement dépendantes du contexte génétique des différents individus. Ils ont identifié plusieurs gènes candidats, dont des gènes non-connus de la maladie et qui pourraient être des cibles potentielles pour le traitement de la maladie chez l’homme, plusieurs des gènes candidats ayant un homologue chez l’homme.
1.6 La levure comme organisme modèle
La levure S. cerevisiae est également un organisme modèle qui peut être utilisé pour comprendre l’effet du contexte génétique et des gènes modificateurs sur les maladies génétiques. Des études telles que celles menées par Ehrenreich et al. (2010) et Hou et al. (2016) ont permis d’établir les bases génétiques derrière des différences phénotypiques entre différentes souches de levure, et ainsi montré que la levure peut elle-même être utilisée pour ce type d’étude (Ehrenreich et al., 2010; Hou et al., 2016). D’autant plus que les différentes caractéristiques de ce petit organisme font de lui un bon organisme modèle. Effectivement, cet organisme a un petit génome d’environ 13 millions de paires de bases (pb), un mode de reproduction par division cellulaire ou par voie sexuée, un temps de génération court et de nombreuses techniques développées pour pouvoir étudier son génome et la fonction de ses gènes (Alberts, 2008; Botstein and Fink, 2011). De plus, malgré que leur ancêtre commun remonte à plus d’un milliard d’années, la levure a plus du tiers de ses gènes qui sont orthologues à ceux de l’humain (Douzery et al., 2004; O'Brien et al., 2005). Cet organisme a donc été utilisé pour pouvoir étudier des gènes humains (Balakrishnan et al., 2012; Botstein and Fink, 2011). En effet, la levure a été utilisée comme modèle pour étudier les déterminants
génétiques des maladies humaines à de multiples reprises depuis le milieu des années 1980 (Jacq, 2004; Kataoka et al., 1985). Ces études avaient comme principe le remplacement du gène de la levure par un gène humain, normal ou muté, pour vérifier par croissance la fonction ou l’effet du gène. Ce type d’expérience a permis d’établir que plusieurs gènes humains peuvent remplacer des gènes chez la levure et y être étudiés (Dolinski and Botstein, 2007). De plus, récemment, des études utilisant des approches à large échelle et des outils de la génomique ont permis de discerner plusieurs gènes humains étant capables de remplacer des gènes essentiels chez la levure (Hamza et al., 2015; Kachroo et al., 2015). Cette possibilité permet donc d’avoir des gènes candidats à étudier dans un organisme plus simple que l’organisme complexe qu’est l’humain.
1.7 Un modèle de levure pour une maladie humaine : le Syndrome de
Wiskott-Aldrich
Dans une étude récente, nous avons utilisé une approche d’évolution expérimentale chez la levure pour cibler des gènes candidats permettant de corriger le phénotype délétère de la maladie humaine du Syndrome de Wiskott-Aldrich (Filteau et al., 2015). Le Syndrome de Wiskott-Aldrich (WAS) est une forme d’immunodéficience primaire rare liée au sexe qui provoque des troubles au niveau des plaquettes sanguines. Les symptômes caractéristiques sont des infections récurrentes, un saignement anormal et de l’eczéma qui peut parfois être très sévère (Sullivan et al., 1994). Selon la sévérité des symptômes, un système de classement de la maladie est utilisé (Notarangelo et al., 2008; Zhu et al., 1995). Pour les patients présentant la maladie classique du WAS, ceux-ci ne dépassent en général pas leur deuxième ou troisième décennie de vie en absence de traitement (Hacein-Bey Abina et al., 2015), avec une médiane d’environ 14.5 années de vie pour des patients étudiés entre 1980 et 2000 (Dupuis-Girod et al., 2003). Cette maladie affecte en général entre 1 et 10 garçons sur 1 million de nouveau-nés (Bosticardo et al., 2009; Buchbinder et al., 2014). Présentement, le traitement privilégié et curatif pour cette maladie est la greffe de cellules souches hématopoïétiques avec un taux de survie d’environ 80% pour les donneurs identiques ou reliés, apparentés ou non, et d’environ 50% pour les donneurs alternatifs. Les donneurs
alternatifs sont utilisés dans les cas où les patients sont hautement classés au niveau de la maladie. Malgré tout, plusieurs complications peuvent survenir suite à ce traitement. Des traitements alternatifs pour chacun des symptômes peuvent également être utilisés en absence de donneur. De nouveaux traitements impliquant des greffes de cellules souches hématopoïétiques génétiquement modifiées avec un virus transporteur du gène désiré pour l’intégrer au génome sont en train d’émerger, ainsi que d’autres traitements potentiels (Buchbinder et al., 2014).
Le WAS est causé par des mutations dans le gène codant pour la protéine d’échafaudage WASP. Cette protéine est notamment impliquée au niveau de l’assemblage du cytosquelette d’actine (Derry et al., 1994; Thrasher and Burns, 2010). Pour les différentes formes de la maladie, plus de 150 mutations uniques ont d’ailleurs été retrouvées (Ochs and Thrasher, 2006). WASP a un homologue fonctionnel chez la levure S. cerevisiae, soit le gène LAS17.
LAS17 est lui aussi connu comme étant un facteur d’assemblage de l’actine (Li, 1997). Son
domaine C-terminal, incluant les domaines WH2 et acidique, active le complexe Arp2/3, responsable de la nucléation des filaments d’actine, et une étude récente suggère que Las17p est également en mesure d’effectuer par elle-même la nucléation à l’aide de sa région riche en proline (Urbanek et al., 2013; Winter et al., 1999). Le domaine WH1, quant à lui, permet la liaison de Las17p avec la Vrp1p. VRP1 est l’homologue du gène WIP codant pour la protéine humaine interagissant avec WASP et est impliqué au niveau de la localisation de Las17p dans la cellule (Lechler et al., 2001). Les mutants du gène LAS17 peuvent avoir différents problèmes au niveau du cytosquelette d’actine et de l’endocytose (Li, 1997; Naqvi
et al., 1998). Des études ont montré qu’en présence de son interactant protéique WIP, WASP
peut compenser la perte de fonction de las17∆ (Rajmohan et al., 2009). Par ailleurs, chez la levure, l’allèle las17-41 provoque un dysfonctionnement de la cellule dans certaines conditions. L’allèle las17-41 de la levure qui contient une mutation W41R est homologue à l’une des mutations provoquant les symptômes classiques de WAS, soit la mutation W64R située au niveau du domaine WH1 de WASP (Figure 1.1) (Fillat et al., 2000; Filteau et al., 2015; Jin et al., 2004). Cet allèle chez la levure cause la thermosensibilité : la levure est en mesure de croitre à une température de 22ºC, mais non à 37ºC (Figure 1.2) (Filteau et al., 2015).
Figure 1.1 Alignement des séquences de WASP et ces homologues.
Le résidu conservé de W64 est montré en gras. Cette figure est tirée de l’article Filteau et al. (2015).
Figure 1.2 Phénotype de thermosensibilité de l’allèle las17-41 à 37ºC.
La croissance des souches BY las17-41-KanMX (BY las17-41) et RM las17-41-KanMX (RM las17-41) à des températures variant de 22ºC à 37ºC permet de montrer la thermosensibilité comparativement aux souches sauvages RM11 ho∆ et BY4741 ho∆ (RM WT haploid et BY WT haploid respectivement). La technique des dilutions en série est utilisée pour montrer le niveau de croissance à différentes concentrations de culture. La croissance est montrée sur des milieux synthétiques contenant du glucose ou du galactose. Cette figure est adaptée et tirée de l’article Filteau et al. (2015).
Ce système nous a permis de retracer des solutions compensatoires permettant de corriger le phénotype de la maladie au niveau génétique et pharmacologique, mais aussi de déterminer que le contexte génétique et l’environnement influencent ces solutions compensatoires (Filteau et al., 2015). Pour ce faire, nous avons utilisé deux sources de carbone, le glucose et le galactose, qui sont utilisées différemment au niveau métabolique chez la levure (Fendt and Sauer, 2010), et deux contextes génétiques, BY4741 las17-41 (BY las17-41-KanMX) et
RM11 las17-41 (RM las17-41-KanMX), dont la provenance représente bien la variabilité génétique chez l’espèce S. cerevisiae (Liti et al., 2009). Pour procéder à l’évolution expérimentale (Filteau et al., 2015), nous avons laissé les cellules évoluer à température permissive (température de la pièce (TP), environ 22°C) pour ensuite les sélectionner à température restrictive (37°C). Cette approche a permis l’accumulation de premières mutations. Certaines de ces mutations ont permis de compenser la perte de fonction de las17-41p à 37ºC. Ces mutations sont nommées des mutations compensatoires et les cellules évoluées, des mutants compensatoires. Un séquençage du génome de plusieurs des mutants obtenus nous a permis d’identifier les gènes touchés par les mutations compensatoires. Parmi toutes les mutations compensatoires retrouvées dans les différents gènes, dont la plupart avaient des liens d’interaction avec la protéine Las17p, certaines laissaient croire à des pertes de fonction totales ou partielles de la protéine, notamment pour les gènes CNB1, BSP1 et
TWF1. Effectivement, dans ces cas, les mutations retrouvées se résumaient à des délétions et
à des insertions de plusieurs paires de bases, ou encore, à des troncations dues à une mutation qui insérait un codon STOP. Parmi ces trois gènes, le cas le moins attendu était celui de CNB1 étant donné que ce gène ne présentait aucun lien d’interaction connu avec LAS17, contrairement à BSP1 et TWF1. CNB1 est plutôt impliqué dans la signalisation Ca2+/calmoduline.
Le gène CNB1 code pour la sous-unité régulatrice de la calcineurine, une protéine phosphatase à sérine/thréonine de régulation Ca2+/calmoduline. Cette protéine phosphatase est un hétérodimère composé de deux sous-unités : la sous-unité régulatrice Cnb1p et une sous-unité catalytique, la calcineurine A (Cnap) sous l’isoforme Cna1p ou Cmp2p (Cyert et
al., 1991; Liu et al., 1991). La calcineurine est impliquée dans plusieurs processus cellulaires
et dans plusieurs voies de transduction des signaux dépendantes du calcium (Rusnak and Mertz, 2000). Elle a aussi un lien avec WASP, l’homologue de Las17p, car lorsque WASP est absente de la cellule, chez les cellules humaines NK, la quantité de calcineurine et de plusieurs autres molécules ayant un lien avec la mobilisation du calcium diminue (Huang et
al., 2005). Ce lien n’a toutefois pas encore été rapporté chez la levure, ce qui en fait un cas
intéressant. Pour vérifier l’effet de l’inactivation du gène CNB1 par les mutations compensatoires, il a été possible d’utiliser la délétion, mais aussi une molécule, la cyclosporine A (CsA) (Filteau et al., 2015). Cette molécule a d’abord été trouvée et utilisée
comme antifongique, mais celle-ci a également montré une activité immunosuppressive, ce qui en a fait un médicament immunosuppresseur notamment utilisé comme traitement pour les greffes d’organes (Tribe, 1998). La CsA s’assemble avec la cyclophiline A (CypA), présente dans les cellules, pour inhiber la calcineurine (Filteau et al., 2015). Ce complexe cible spécifiquement une région entre la sous-unité régulatrice Cnb1p et la sous-unité catalytique Cnap de la calcineurine (Cardenas et al., 1995; Huai et al., 2002). Nous avons testé expérimentalement l’inhibition de la calcineurine, et donc Cnb1p, par ce médicament pour vérifier s’il permettait la correction de l’effet de la perte de fonction de las17-41p à 37ºC (Filteau et al., 2015). L’hypothèse a été confirmée : il y avait correction dans au moins une des conditions et plus spécifiquement, la réponse (ou la correction) était différente dépendamment du contexte génétique dans un environnement donné, soit BY
las17-41-KanMX en glucose (Figure 1.3). Cette spécificité montre une différence au niveau du génome
qui permet de supporter encore une fois que la variabilité génétique influence la réponse à un traitement donné. Comme l’effet du médicament est dépendant du contexte génétique, ce système de correction d’une mutation délétère par inhibition d’un gène à l’aide d’un médicament est un modèle idéal pour comprendre comment cette dépendance prend place au niveau moléculaire.
Figure 1.3 Correction du phénotype dû à la perte de fonction de las17-41 à 37ºC par l’inhibition de la calcineurine par la cyclosporine A (CsA) à une concentration de 10 µg/ml.
Comme précédemment mentionné, la CsA permet l’inhibition de la calcineurine en ciblant une région entre la sous-unité régulatrice Cnb1p et la sous-unité catalytique Cnap. Cette inhibition permet une correction spécifique au contexte génétique de BY las17-41-KanMX (BY las17-41). Cette figure est tirée de l’article Filteau et al. (2015).
1.8 Objectifs et hypothèses
Le modèle présenté est à la fois représentatif d’un traitement personnalisé pour une maladie et montre des différences de réponse au traitement dépendamment du contexte génétique. Ce modèle est donc idéal pour arriver à comprendre le fonctionnement des effets du contexte génétique sur la réponse à un médicament du point de vue moléculaire. Mon objectif principal est donc d’identifier les bases génétiques et moléculaires qui influencent la différence de réponse à un traitement, la CsA, entre les deux contextes génétiques utilisés, BY
las17-41-KanMX et RM las17-41-las17-41-KanMX, qui ont une divergence d’environ 0.5% au niveau de leur
génome (Ruderfer et al., 2006). Plus spécifiquement, mon objectif est d’identifier les déterminants génétiques responsables de cette réponse dépendante au contexte génétique.
Comme les mutations de l’allèle las17-41 causent une perte de fonction de la protéine à 37°C, suggérée par la thermosensibilité de la souche à cette même température, la première hypothèse que nous avons posée est que l’abondance de la protéine est probablement affectée. En effet, étant donné que la protéine n’est plus en mesure d’accomplir sa fonction, potentiellement causé par un mauvais repliement, celle-ci est probablement dégradée rapidement au sein de la cellule, ou encore même, tout simplement non exprimée (Jin et al., 2004; Williamson, 2012). Ainsi, pour restaurer la croissance à 37°C, la CsA pourrait avoir un impact direct sur la protéine. En effet, le médicament pourrait permettre de stabiliser la protéine las17-41p si elle est produite, donnant lieu à une durée de vie plus longue dans la cellule et ainsi, celle-ci pourrait accomplir sa fonction dans le contexte génétique de BY. Cet impact pourrait donc nous donner des pistes sur les gènes modificateurs responsables de la différence de réponse entre les deux contextes génétiques.
Une approche plus ciblée a également été utilisée pour arriver à identifier directement les gènes modificateurs responsables de la différence de réponse. L’approche utilisée pour répondre à cet objectif a été le croisement des deux contextes génétiques, puis l’identification de gènes candidats possiblement responsables de la variation de réponse par analyse de type QTL (locus de caractères quantitatifs), soit analyse de ségrégants en pool (« Bulk segregant analysis »), à partir des hybrides F1 (Ehrenreich et al., 2009; Ehrenreich et al., 2010; Meijnen
et al., 2016; Swinnen et al., 2012). Cette approche a d’ailleurs déjà été utilisée dans plusieurs
études pour comprendre les bases moléculaires qui influencent la différence phénotypique entre deux souches de levures dans différentes conditions. Notamment, Meijnen et al. (2016) se sont servis de cette approche pour discerner les traits qui influencent la haute tolérance à l’acide acétique alors que Swinnen et al. (2012) ont identifié les bases moléculaires derrière une grande tolérance à l’éthanol.
Cette approche consiste plus précisément à obtenir différents ségrégants avec le génome recombinant des parents. Ainsi, dépendamment de la distribution des régions des deux génomes chez les ségrégants, ceux-ci auront différents phénotypes de réponse à la CsA avec certains se rapprochant de BY KanMX et d’autres se rapprochant de RM
las17-41-KanMX. Le patron de réponse du croisement en lui-même donnera un indice sur la dominance
patron de réponse de chacune des spores donnera, quant à lui, un indice sur le nombre de gènes impliqués. Suite à un séquençage des deux pools de phénotypes opposés, un ou plusieurs gènes candidats de la différence de réponse à la CsA pourront être identifiés par analyse statistique de ségrégants en pool. Notre hypothèse est que, comme démontré dans notre étude d’évolution expérimentale, les gènes candidats seront des gènes impliqués dans le réseau d’interaction de LAS17 ou de CNB1, ou encore à l’interface entre les deux gènes (Filteau et al., 2015). Finalement, différentes approches ont été utilisées pour essayer de confirmer l’identification des gènes candidats dans le contexte génétique répondant à la CsA.
Chapitre 2 - Matériel et méthodes
2.1 Milieux de culture
Le milieu complexe (YPD) utilisé est composé de 1% d’extrait de levure, 2% de tryptone et 2% de glucose. Le milieu synthétique complet (Sc) utilisé est composé de 0.175% de base d’azote de levure, 2% de glucose, 0.1% de glutamate monosodique (MSG), et 0.135% de drop out complet, 0.127% de drop out –ura, 0.097% de drop out –leu ou 0.127% de drop out –lys. Le milieu Sc –met utilisé est plutôt composé de 0.175% de base d’azote de levure, 2% de glucose, 0.5% d’ammonium sulfate et 0.135% de drop out –met. Un 2% d’agar est ajouté pour obtenir un milieu solide. Les antibiotiques suivants ont été ajoutés selon les besoins à ces concentrations finales : 200 µg/ml de généticine (G418) (Bioshop, Burlington, Ontario, Canada), 100 µg/ml de nourseothricine (Nat) (WERNER BioAgents, Iéna, Allemagne) et 250 µg/ml d’hygromycine B (Hyg) (Bioshop). Lorsque le milieu Sc est supplémenté d’Hyg, de l’acide succinique à une concentration finale de 0.05 M est ajouté et le pH du milieu est par la suite ajusté à 6 à l’aide de NaOH avant le passage à l’autoclave.
Le MSG a été utilisé comme source d’azote pour la composition du Sc étant donné que Cheng
et al. (2000) ont rapporté une modification de l’efficacité de la sélection du G418 en présence
d’ammonium sulfate (Cheng et al., 2000). De plus, ce milieu a été utilisé dans l’article de Filteau et al. (2015). La sélection du milieu Sc –met avec MSG étant altérée, une exception a été faite pour ce milieu.
2.2 Souches
Les souches utilisées lors de cette étude, soit BY las17-41 et RM las17-41, sont tirées directement de l’article Filteau et al. (2015). Dans le cadre de la présente étude, celles-ci seront nommées BY las17-41-KanMX et RM las17-41-KanMX. Ces deux cas représentent respectivement les contextes génétiques de BY4741 (BY) et RM11-1a (RM), deux souches
de Saccharomyces cerevisiae ayant une divergence d’environ 0.5% au niveau de leur génome (Ruderfer et al., 2006). Les souches thermosensibles avec l’allèle de las17-41 ont toutes été obtenues à partir de ces deux souches et quelques manipulations génétiques (Tableau 2.1).
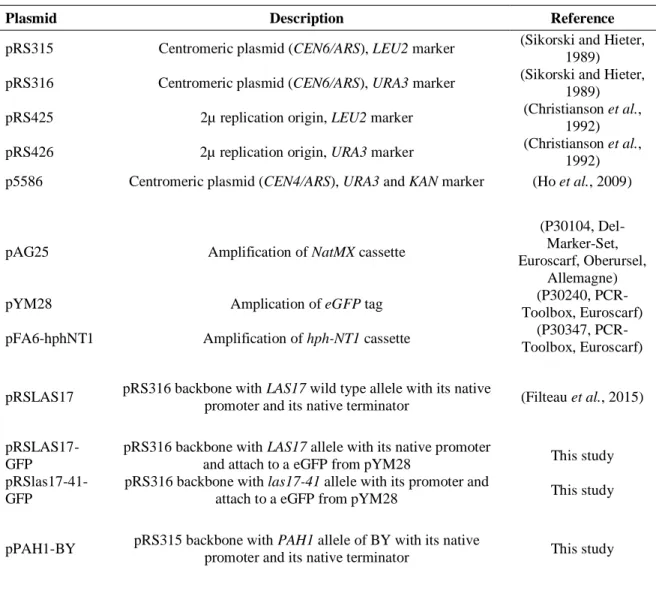
Tableau 2.1 Souches de S. cerevisiae utilisées et construites
Strain Genotype Plasmid Reference
BY las17-41-KanMX MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ho
las17-41-KAN None (Filteau et al., 2015)
RM las17-41-KanMX MATa leu2Δ0 ura3Δ0 ho::loxp
amn1-A1103T las17-41-KAN None (Filteau et al., 2015)
BY4741 ho∆ MATa his3∆1 leu2∆0 met15 ura3∆0
ho::KANMX None (Giaever et al., 2002)
RM11 ho∆ MATa leu2Δ0 ura3Δ0 ho::KANr
amn1-A1103T None
Original name of the strain is YEF1946. (Yvert et al., 2003) BY las17∆ MATα his3Δ1 leu2Δ0 lys2Δ0 met15Δ0
ura3Δ0 ho las17::KANMX None (Filteau et al., 2015)
BY4742 MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 ho None (Open Biosystems, Canada) BY4742 pRS316 MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 ho pRS316 This study
BY las17-41-NatMX MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ho
las17-41-NAT
With and without pRSLAS17
This study
RM las17-41-NatMX MATa leu2Δ0 ura3Δ0 ho::loxp
amn1-A1103T las17-41-NAT
With and without pRSLAS17
This study
BY4741 MATa his3∆1 leu2∆0 met15∆0 ura3∆0 ho None (Open Biosystems)
BYxRM
LAS17/las17-41-KanMX pRS316
MATα/MATa his3∆1/HIS3 leu2∆0/leu2∆0
lys2∆0/LYS2 ura3∆0/ura3∆0 ho/ho::loxp AMN1/amn1-A1103T LAS17/las17-41-KAN
pRS316 This study
BY las17-41-NatMX
app1∆ MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ho las17-41-NAT app1-HPH
With and without pRSLAS17
This study RM las17-41-NatMX
app1∆ MATa leu2Δ0 ura3Δ0 ho::loxp amn1-A1103T las17-41-NAT app1-HPH
With and without pRSLAS17
This study BY las17-41-NatMX
end3∆ MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ho las17-41-NAT end3-HPH pRSLAS17 This study
RM las17-41-NatMX
end3∆
MATa leu2Δ0 ura3Δ0 ho::loxp
BY las17-41-NatMX
mkt1∆ MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ho las17-41-NAT mkt1-HPH
With and without pRSLAS17
This study RM las17-41-NatMX
mkt1∆ MATa leu2Δ0 ura3Δ0 ho::loxp amn1-A1103T las17-41-NAT mkt1-HPH
With and without pRSLAS17
This study BY las17-41-NatMX
nst1∆ MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ho las17-41-NAT nst1-HPH
With and without pRSLAS17
This study RM las17-41-NatMX
nst1∆ MATa leu2Δ0 ura3Δ0 ho::loxp amn1-A1103T las17-41-NAT nst1-HPH
With and without pRSLAS17
This study BY las17-41-NatMX
pah1∆ MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ho las17-41-NAT pah1-HPH pRSLAS17 This study
RM las17-41-NatMX
pah1∆ MATa leu2Δ0 ura3Δ0 ho::loxp amn1-A1103T las17-41-NAT pah1-HPH pRSLAS17 This study
BY LAS17/las17-41
MATα/a his3Δ1/his3Δ1 leu2Δ0/leu2Δ0
lys2Δ0/LYS2 MET15/met15Δ0
ura3Δ0/ura3Δ0 ho/ho LAS17/las17-41-KAN None (Filteau et al., 2015)
BY las17-41-KanMX MATα
MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ho
las17-41-KAN None This study
RM LAS17/las17-41
MATα/a ho::URA/ho::loxp leu2Δ0/leu2Δ0
ura3Δ0/ura3Δ0 amn1-A1103T/amn1-A1103T LAS17/las17-41-KAN
None (Filteau et al., 2015) RM las17-41-KanMX
MATα
MATα ho::loxp leu2Δ0 ura3Δ0
amn1-A1103T las17-41-KAN None This study
BYxRM
las17-41- KanMX/las17-41-NatMX
MATα/a his3∆1/HIS3 leu2∆0/leu2Δ0
lys2∆0/LYS2 ura3Δ0/ura3∆0 ho/ho::loxp AMN1/amn1-A1103T las17-41-KAN/las17-41-NAT With and without pRSLAS17 This study RMxBY las17-41- KanMX/las17-41-NatMX
MATα/a HIS3/his3∆1 leu2∆0/leu2Δ0
MET15/met15∆0 ura3Δ0/ura3∆0 ho::loxp/ho amn1-A1103T/AMN1 las17-41-KAN/las17-41-NAT With and without pRSLAS17 This study BYxBY las17-41- KanMX/las17-41-NatMX
MATα/a his3∆1/his3∆1 leu2∆0/leu2Δ0
lys2∆0/LYS2 MET15/met15∆0 ura3Δ0/ura3∆0 ho/ho
las17-41-KAN/las17-41-NAT
None This study RMxRM
las17-41- KanMX/las17-41-NatMX
MATα/a ho::loxp/ho::loxp leu2∆0/leu2Δ0
ura3Δ0/ura3∆0 amn1-A1103T/amn1-A1103T las17-41-KAN/las17-41-NAT
None This study BYxRM
las17-41- KanMX/las17-41-NatMX APP1/app1
MATα/a his3∆1/HIS3 leu2∆0/leu2Δ0
lys2∆0/LYS2 ura3Δ0/ura3∆0 ho/ho::loxp AMN1/amn1-A1103T las17-41-KAN/las17-41-NAT APP1-BY/app1-HPH With and without pRSLAS17 This study RMxBY las17-41- KanMX/las17-41-NatMX APP1/app1
MATα/a HIS3/his3∆1 leu2∆0/leu2Δ0
MET15/met15∆0 ura3Δ0/ura3∆0 ho::loxp/ho amn1-A1103T/AMN1 las17-41-KAN/las17-41-NAT APP1-RM/app1-HPH With and without pRSLAS17 This study BYxBY las17-41- KanMX/las17-41-NatMX APP1/app1
MATα/a his3∆1/his3∆1 leu2∆0/leu2Δ0
ura3Δ0/ura3∆0 ho/ho las17-41-KAN/las17-41-NAT APP1/app1-HPH
RMxRM
las17-41- KanMX/las17-41-NatMX APP1/app1
MATα/a ho::loxp/ho::loxp leu2∆0/leu2Δ0
ura3Δ0/ura3∆0 amn1-A1103T/amn1-A1103T las17-41-KAN/las17-41-NAT
APP1/app1-HPH
None This study
BYxRM
las17-41- KanMX/las17-41-NatMX END3/end3
MATα/a his3∆1/HIS3 leu2∆0/leu2Δ0
lys2∆0/LYS2 ura3Δ0/ura3∆0 ho/ho::loxp AMN1/amn1-A1103T las17-41-KAN/las17-41-NAT END3-BY/end3-HPH With and without pRSLAS17 This study RMxBY las17-41- KanMX/las17-41-NatMX END3/end3
MATα/a HIS3/his3∆1 leu2∆0/leu2Δ0
MET15/met15∆0 ura3Δ0/ura3∆0 ho::loxp/ho amn1-A1103T/AMN1 las17-41-KAN/las17-41-NAT END3-RM/end3-HPH With and without pRSLAS17 This study BYxBY las17-41- KanMX/las17-41-NatMX END3/end3
MATα/a his3∆1/his3∆1 leu2∆0/leu2Δ0
lys2∆0/LYS2 MET15/met15∆0 ura3Δ0/ura3∆0 ho/ho
las17-41-KAN/las17-41-NAT END3/end3-HPH
None This study
RMxRM
las17-41- KanMX/las17-41-NatMX END3/end3
MATα/a ho::loxp/ho::loxp leu2∆0/leu2Δ0
ura3Δ0/ura3∆0 amn1-A1103T/amn1-A1103T las17-41-KAN/las17-41-NAT
END3/end3-HPH
None This study
BYxRM
las17-41- KanMX/las17-41-NatMX MKT1/mkt1
MATα/a his3∆1/HIS3 leu2∆0/leu2Δ0
lys2∆0/LYS2 ura3Δ0/ura3∆0 ho/ho::loxp AMN1/amn1-A1103T las17-41-KAN/las17-41-NAT MKT1-BY/mkt1-HPH With and without pRSLAS17 This study RMxBY las17-41- KanMX/las17-41-NatMX MKT1/mkt1
MATα/a HIS3/his3∆1 leu2∆0/leu2Δ0
MET15/met15∆0 ura3Δ0/ura3∆0 ho::loxp/ho amn1-A1103T/AMN1 las17-41-KAN/las17-41-NAT MKT1-RM/mkt1-HPH With and without pRSLAS17 This study BYxBY las17-41- KanMX/las17-41-NatMX MKT1/mkt1
MATα/a his3∆1/his3∆1 leu2∆0/leu2Δ0
lys2∆0/LYS2 MET15/met15∆0 ura3Δ0/ura3∆0 ho/ho
las17-41-KAN/las17-41-NAT MKT1/mkt1-HPH
None This study
RMxRM
las17-41- KanMX/las17-41-NatMX MKT1/mkt1
MATα/a ho::loxp/ho::loxp leu2∆0/leu2Δ0
ura3Δ0/ura3∆0 amn1-A1103T/amn1-A1103T las17-41-KAN/las17-41-NAT
MKT1/mkt1-HPH
None This study
BYxRM
las17-41- KanMX/las17-41-NatMX NST1/nst1
MATα/a his3∆1/HIS3 leu2∆0/leu2Δ0
lys2∆0/LYS2 ura3Δ0/ura3∆0 ho/ho::loxp AMN1/amn1-A1103T las17-41-KAN/las17-41-NAT NST1-BY/nst1-HPH With and without pRSLAS17 This study RMxBY las17-41- KanMX/las17-41-NatMX NST1/nst1
MATα/a HIS3/his3∆1 leu2∆0/leu2Δ0
MET15/met15∆0 ura3Δ0/ura3∆0 ho::loxp/ho amn1-A1103T/AMN1 las17-41-KAN/las17-41-NAT NST1-RM/nst1-HPH With and without pRSLAS17 This study BYxBY las17-41- KanMX/las17-41-NatMX NST1/nst1
MATα/a his3∆1/his3∆1 leu2∆0/leu2Δ0
lys2∆0/LYS2 MET15/met15∆0 ura3Δ0/ura3∆0 ho/ho
las17-41-KAN/las17-41-NAT NST1/nst1-HPH
None This study
RMxRM
las17-41- KanMX/las17-41-NatMX NST1/nst1
MATα/a ho::loxp/ho::loxp leu2∆0/leu2Δ0
ura3Δ0/ura3∆0 amn1-A1103T/amn1-A1103T las17-41-KAN/las17-41-NAT
NST1/nst1-HPH
BYxRM
las17-41- KanMX/las17-41-NatMX PAH1/pah1
MATα/a his3∆1/HIS3 leu2∆0/leu2Δ0
lys2∆0/LYS2 ura3Δ0/ura3∆0 ho/ho::loxp AMN1/amn1-A1103T las17-41-KAN/las17-41-NAT PAH1-BY/pah1-HPH With and without pRSLAS17 This study RMxBY las17-41- KanMX/las17-41-NatMX PAH1/pah1
MATα/a HIS3/his3∆1 leu2∆0/leu2Δ0
MET15/met15∆0 ura3Δ0/ura3∆0 ho::loxp/ho amn1-A1103T/AMN1 las17-41-KAN/las17-41-NAT PAH1-RM/pah1-HPH With and without pRSLAS17 This study BYxBY las17-41- KanMX/las17-41-NatMX PAH1/pah1
MATα/a his3∆1/his3∆1 leu2∆0/leu2Δ0
lys2∆0/LYS2 MET15/met15∆0 ura3Δ0/ura3∆0 ho/ho
las17-41-KAN/las17-41-NAT PAH1/pah1-HPH
None This study
RMxRM
las17-41- KanMX/las17-41-NatMX PAH1/pah1
MATα/a ho::loxp/ho::loxp leu2∆0/leu2Δ0
ura3Δ0/ura3∆0 amn1-A1103T/amn1-A1103T las17-41-KAN/las17-41-NAT
PAH1/pah1-HPH
None This study
BYxRM
las17-41- KanMX/las17-41-NatMX SWP1/swp1
MATα/a his3∆1/HIS3 leu2∆0/leu2Δ0
lys2∆0/LYS2 ura3Δ0/ura3∆0 ho/ho::loxp AMN1/amn1-A1103T las17-41-KAN/las17-41-NAT SWP1-BY/swp1-HPH With and without pRSLAS17 This study RMxBY las17-41- NatMX/las17-41-KanMX SWP1/swp1
MATa/α HIS3/his3∆1 leu2∆0/leu2Δ0
LYS2/lys2∆0 ura3Δ0/ura3∆0 ho::loxp/ho amn1-A1103T/AMN1 las17-41-NAT/las17-41-KAN SWP1-RM/swp1-HPH With and without pRSLAS17 This study BY las17-41-NatMX END3-BY
MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ho
las17-41-NAT END3-BY None This study
BY las17-41-NatMX
END3-BY S258N
MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ho
las17-41-NAT END3-BY S258N None This study
BY las17-41-NatMX
END3-BY S258D
MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ho
las17-41-NAT END3-BY S258D None This study
BY las17-41-NatMX
END3-BY S258A
MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ho
las17-41-NAT END3-BY S258A None This study
BY las17-41-NatMX
END3-BY D268N
MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ho
las17-41-NAT END3-BY D268N None This study
BY las17-41-NatMX
END3-RM
MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ho
las17-41-NAT END3-RM None This study
RM las17-41-NatMX
END3-BY
MATa leu2Δ0 ura3Δ0 ho::loxp
amn1-A1103T las17-41-NAT END3-BY None This study
RM las17-41-NatMX
END3-BY 2x
MATa leu2Δ0 ura3Δ0 ho::loxp
amn1-A1103T las17-41-NAT END3-BY 2x None This study
RM las17-41-NatMX
END3-RM
MATa leu2Δ0 ura3Δ0 ho::loxp
amn1-A1103T las17-41-NAT END3-RM None This study ho allele of BY genetic background : recessive allele with amino acid change T189A, G223S, L405S, H475L
(Meiron et al., 1995)
Comme certaines méthodes décrites plus bas nécessitaient l’utilisation d’un marqueur de résistance à la Kanamycine (Kan) et au G418, la cassette KanMX des souches BY
las17-41-KanMX et RM las17-41-las17-41-KanMX a été modifiée pour une cassette de résistance au Nat : NatMX (Tableau 2.1). Au préalable, pour éviter des problèmes causés par le nombre de
manipulations génétiques à effectuer avec ces souches, les souches BY las17-41-KanMX et RM las17-41-KanMX ont été transformées directement avec le plasmide pRSLAS17 permettant de restaurer la fonction de Las17p (Filteau et al., 2015). Trois colonies isolées de chaque souche ont été conservées pour les étapes subséquentes. La cassette KanMX a été remplacée par la cassette NatMX, préalablement amplifiée du plasmide pAG25 (Tableau 2.2 pour le plasmide; Tableau 2.3 pour les amorces de construction et de confirmation) et digérée avec l’enzyme DpnI (New England Biolabs (NEB), Ipswich, Massachusetts, États-Unis), en suivant une procédure standard de transformation utilisant des cellules compétentes (Güldener et al., 1996). Les colonies transformées ont été sélectionnées sur milieu Sc –ura +Nat.
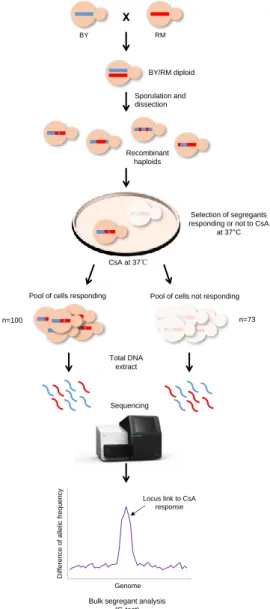
Tableau 2.2 Plasmides utilisés et construits
Plasmid Description Reference
pRS315 Centromeric plasmid (CEN6/ARS), LEU2 marker (Sikorski and Hieter, 1989) pRS316 Centromeric plasmid (CEN6/ARS), URA3 marker (Sikorski and Hieter,
1989) pRS425 2µ replication origin, LEU2 marker (Christianson et al.,
1992) pRS426 2µ replication origin, URA3 marker (Christianson et al.,
1992) p5586 Centromeric plasmid (CEN4/ARS), URA3 and KAN marker (Ho et al., 2009)
pAG25 Amplification of NatMX cassette
(P30104, Del-Marker-Set, Euroscarf, Oberursel,
Allemagne)
pYM28 Amplication of eGFP tag (P30240,
PCR-Toolbox, Euroscarf) pFA6-hphNT1 Amplification of hph-NT1 cassette (P30347,
PCR-Toolbox, Euroscarf)
pRSLAS17 pRS316 backbone with LAS17 wild type allele with its native
promoter and its native terminator (Filteau et al., 2015)
pRSLAS17-GFP
pRS316 backbone with LAS17 allele with its native promoter
and attach to a eGFP from pYM28 This study
pRSlas17-41-GFP
pRS316 backbone with las17-41 allele with its promoter and
attach to a eGFP from pYM28 This study
pPAH1-BY pRS315 backbone with PAH1 allele of BY with its native