CHAPITE II
Oxydation du méthane à basse température
Methane oxidation at low temperature
Oxydation du méthane à haute température
Methane oxidation at high temperature
THESE
Pour l’obtention du grade de
DOCTEUR de l’UNIVERSITE de POITIERS (DIPLÔME National – Arrêté du 7 Août 2006)
Ecole Doctorale Ingénierie Chimique, Biologique et Géologique Secteur de Recherche : Chimie Appliquée
Présentée par Alexandre BAYLET
INGENIEUR en CHIMIE et PHYSIQUE (ENSCPB)
OXYDATION du METHANE à BASSE et HAUTE TEMPERATURE.
APPLICATION DE PROCEDES PLASMA et/ou CATALYSE.
Directeur de thèse : Daniel DUPREZ Co-directeur de thèse : Patrice MARECOT
Soutenance prévue le 26 SEPTEMBRE 2008 devant la Commission d’Examen JURY
Rapporteurs :
Gianpiero GROPPI Professeur, Politecnico Milano, Italie Antonio GUERRERO RUIZ Professeur, UNED-ICP, Espagne Stéphane PASQUIERS Directeur de recherche, LPGP, Orsay Examinateurs:
Patrick GELIN Directeur de recherche, IRCE, Lyon Daniel DUPREZ Directeur de recherche, LACCO, Poitiers Patrice MARECOT Directeur de recherche, LACCO, Poitiers Jean-Michel TATIBOUËT Directeur de recherche, LACCO, Poitiers
Xavier JEANDEL Ingénieur de Recherches, Renault, Technocentre, Guyancourt
DOCTOR COMMUNITATIS
Remerciements
Ces travaux ont été réalisés au sein du Laboratoire de Catalyse en Chimie Organique (LACCO, UMR-CNRS 6503) dans le cadre d’une thèse cofinancée par Renault SAS et l’Agence De l’Environnement et de la Maîtrise de l’Energie (ADEME).
Je tiens à remercier Mr Daniel Duprez, Directeur de thèse et Directeur de Recherche CNRS de m’avoir accueilli au sein du LACCO ainsi que Mr Patrice Marécot et Mr Jean-Michel
Tatibouët, Directeurs de Recherche CNRS et co-Directeurs de thèse et Mr Sébastien Royer, maître
de conférence, pour les débats très passionnés sur la catalyse.
Je tiens également à remercier Mme Karine Lombaert et Mr Xavier Jeandel, tuteurs Renault SAS de la thèse, pour m’avoir confié ce sujet très intéressant sur le traitement du méthane, molécule ayant une part importante dans le réchauffement climatique, pour m’avoir permis de travailler sur le développement d’une nouvelle technologie de dépollution automobile et pour leur suivi de ce travail, leur disponibilité et leur sympathie.
Je remercie également Mr Patrice Gelin, Directeur de Recherche CNRS à l’IRCE Lyon, et Mr Stéphane Pasquiers, Directeur de recherche au LPGP Paris, pour avoir accepté d’être membres du jury et rapporteurs de ce travail, le Professeur Antonio Guerrero Ruiz pour avoir accepté d’être rapporteur, ainsi que Mr Stéphane Barbusse, Ingénieur ADEME.
Je voulais aussi remercier le Professeur Pio Forzatti qui m’a donné l’opportunité d’effectuer un stage d’une période de trois mois au Politecnico di Milano, ainsi que le Professeur Gianpiero
Groppi qui m’a confié un sujet très intéressant sur le palladium, qui reste toujours un matériau
« étrange », et qui a partagé son temps pour la discussion et la compréhension des résultats. Je souhaite remercier également Mlle Paola Castellazzi, doctorante et Marco Schioppetti, élève au Politecnico pour leur collaboration dans ce travail et les autres membres de l’équipe pour leur accueil et leur sympathie.
Mes remerciements s’adressent également à Danielle Mesnard, Elisabeth Colnay, Sandrine
Arrii-Clacens, Mehrad Tarighi, Stéphane Pronier pour la caractérisation des catalyseurs et à Michel Chauveau, Jean-Jacques Colin et Tristan Beldi, pour leurs spécificités techniques
indispensables.
Je remercie également Aurélie Vicente et Crina Corbos, doctorantes, Angélique Bosdeveix, ancienne DEPSUP, et tous les collègues de l’équipe Catalyse par les Métaux.
MERCI à ma Famille pour son soutien.
Alexandre BAYLET Poitiers, Juillet 2008
CHAPITRE I: TECHNOLOGIE PLASMA/CATALYSE ...6
I.1. PARTIE BIBLIOGRAPHIQUE ...6
I.1.1. LE PLASMA...6
I.1.2. PLASMA NON-THERMIQUE...6
I.1.3. PLASMA THERMIQUE...8
I.1.4. CONFIGURATIONS TECHNOLOGIQUES...11
I.1.4.1. Configurations de décharges...11
I.1.4.2. Configuration plasma/catalyseur ...11
I.1.5. REACTION DU METHANE DANS UN PLASMA D’AIR...12
I.1.6. RESULTATS EXPERIMENTAUX LIES A L’OXYDATION DU METHANE...12
I.1.6.1. Influence du plasma sur le CH4 seul...12
I.1.6.2. Influence du plasma sur un mélange CH4/O2...13
I.1.6.3. Influence du plasma sur un mélange CH4/H2O ...14
I.1.6.4. Influence du plasma sur un mélange CH4/CO2...15
I.1.6.5. Influence du plasma sur un mélange CH4/O2/H2O...16
I.1.6.6. Influence du plasma sur un mélange CH4/O2/CO2/NO ...16
I.1.6.7. Remarque sur la formation d’ozone ...17
I.1.7. CONCLUSION...17
I.2. PARTIE EXPERIMENTALE GENERALE ...20
I.2.1. MONTAGE D’OXYDATION...20
I.2.2. ANALYSES CHIMIQUES ET PHYSICO-CHIMIQUES...20
I.2.2.1. Diffraction des rayons X...20
I.2.2.2. Analyses des produits de réaction ...20
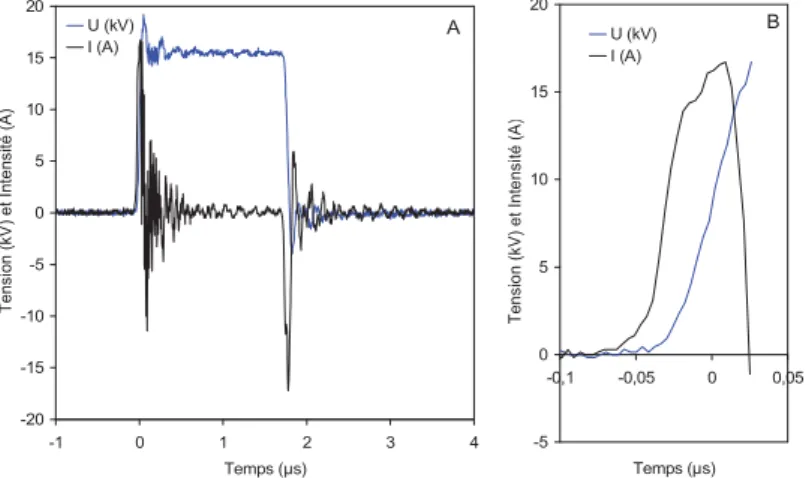
I.2.3. MESURE DES COMPOSANTES ELECTRIQUES : TENSION U(T) ET INTENSITE I(T) ...21
I.2.3.1. Oscilloscope numérique...21
I.2.3.2. Sonde haute tension et sonde d’induction ...21
I.2.4. CALCUL DE PUISSANCE...22
I.2.4.1. Alimentation continue...22
I.2.4.2. Alimentation sinusoïdale ...22
I.2.4.2.1. La méthode du condensateur de Manley ...22
I.2.4.2.2. Les phases d’un plasma ...24
I.2.4.3. Alimentation impulsionnelle ...24
I.3. PLASMA NON-THERMIQUE : RESULTATS ET DISCUSSIONS...25
I.3.1. CONFIGURATION DBD : REACTEUR COAXIAL, ALIMENTATION IMPULSIONNELLE...25
I.3.1.1. Introduction ...25
I.3.1.2. Partie expérimentale...25
I.3.1.2.1. Schéma du montage...25
I.3.1.2.2. Préparation du catalyseur LACCO ...26
I.3.1.3. Effet du plasma seul...26
I.3.1.4. Etude de l’interaction plasma/catalyse ...29
I.3.1.4.1. Remarque : modification du réacteur ...29
I.3.1.4.2. Influence de la position du catalyseur (POST ou IN-plasma) ...30
I.3.1.4.3. Température d’activation du catalyseur...33
I.3.1.5. Conclusion ...34
I.3.2. CONFIGURATION DBD : REACTEUR COAXIAL, ALIMENTATION SINUSOÏDALE...35
I.3.2.1. Introduction ...35
I.3.2.2.1. Schéma du montage et caractéristiques des réacteurs ...35
I.3.2.2.2. Compositions et débits des mélanges gazeux...35
I.3.2.3. Réacteur quartz...36
I.3.2.3.1. Comportement sous mélange N2/O2...36
I.3.2.3.2. Oxydation du CH4...37
I.3.2.3.3. Influence de H2O, du CO2 et du volume plasma ...38
I.3.2.3.3.1. Influence de H2O et du CO2...38
I.3.2.3.3.2. Influence du volume plasma...38
I.3.2.3.4. Comparaison entre alimentation impulsionnelle et alimentation sinusoïdale ...39
I.3.2.3.5. Présence d’un solide IN-plasma ...40
I.3.2.3.5.1. Effet de billes de Al2O3 imprégnées ou non à 1%Pd...40
I.3.2.3.5.2. Effet d’un matériau ferroélectrique BaTiO3...40
I.3.2.4. Réacteur céramique ...46
I.3.2.4.1. Phénomène parasite dû à la conception du réacteur ...47
I.3.2.4.2. Oxydation du CH4 après modification du réacteur...47
I.3.2.4.3. Conditions expérimentales...47
I.3.2.4.5. Résultats...48
I.3.2.4.6. Remarques ...50
I.3.2.5. Conclusion ...51
I.3.3. CONFIGURATION DBD : REACTEUR PLAN/PLAN, ALIMENTATION SINUSOÏDALE...52
I.3.3.1. Introduction ...52
I.3.3.2. Partie expérimentale...52
I.3.3.3. Résultats...52
I.3.3.4. Remarques ...53
I.3.3.5. Conclusion ...54
I.3.4. CONFIGURATION POINTE/PLAN : ALIMENTATION CONTINUE...55
I.3.4.1. Introduction ...55
I.3.4.2. Partie expérimentale...55
I.3.4.3. Résultats et conclusion ...55
I.3.5. CONFIGURATION MULTIPOINTES/PLAN : ALIMENTATION IMPULSIONNELLE...56
I.3.5.1. Introduction ...56
I.3.5.2. Partie expérimentale...56
I.3.5.3. Résultats...56
I.3.5.4. Conclusion ...57
I.4. PLASMA THERMIQUE : RESULTATS ET DISCUSSIONS...58
I.4.1. CONFIGURATION POINTE/PLAN : ALIMENTATION SINUSOÏDALE...58
I.4.1.1. Introduction ...58
I.4.1.2. Partie expérimentale...58
I.4.1.3. Résultats...60
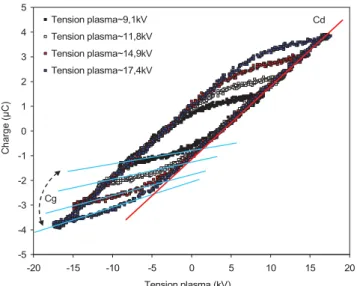
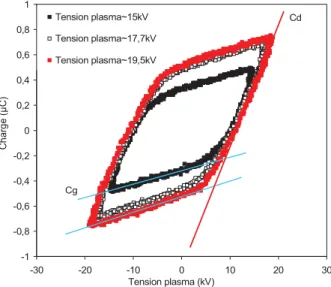
I.4.1.3.1. Etude énergétique ...60
I.4.1.3.1.1. Détermination de la chaleur de combustion ...60
I.4.1.3.1.2. Température adiabatique ...60
I.4.1.3.2. Influence de la composition du mélange gazeux...61
I.4.1.3.2.1. Variation de la concentration initiale en CH4...61
I.4.1.3.2.2. Présence de C3H6 dans le mélange gazeux ...62
I.4.1.3.3. Influence du gap entre les électrodes...63
I.4.1.3.3.1. Conversion du CH4...63
I.4.1.3.3.2. Mesure de température ...64
I.4.1.3.4. Sens de circulation du gaz par rapport à l’arc électrique...66
I.4.1.3.5. Utilisation d’un catalyseur en position POST-plasma...66
I.4.1.3.6. Isolation thermique du réacteur ...68
I.4.1.3.7. Contraintes liées à la génération d’un plasma thermique ...68
I.4.1.3.7.1. Paramètres électriques ...68
I.4.1.3.7.2. Produits de réaction : O3, CO et NOx...69
I.4.1.3.8. Conclusion ...69
I.4.2. CONFIGURATION POINTE/PLAN : ALIMENTATION IMPULSIONNELLE...71
I.4.2.1. Introduction ...71
I.4.2.2. Partie expérimentale...71
I.4.2.2.1. Générateur impulsionnel...71
I.4.2.2.2. Montage plasma...71
I.4.2.3. Résultats...72
I.4.2.3.1. Puissance consommée par le plasma ...72
I.4.2.3.2. Conversion du méthane ...73
I.4.2.4. Conclusion ...74
I.5. CONCLUSION: COMPARAISON DES DIFFERENTES SYSTEMES PLASMA/CATALYSE...75
I.6. REFERENCES BIBLIOGRAPHIQUES ...78
CHAPITRE II : OXYDATION DU METHANE A BASSE TEMPERATURE - METHANE OXIDATION AT LOW TEMPERATURE ...79
II.1. RESUME DE L’ENSEMBLE DE L’ETUDE ...79
II.2. EFFECT OF PD LOADING AND DISPERSION ON REDOX BEHAVIOUR AND CH4 COMBUSTION ACTIVITY OF AL2O3 SUPPORTED CATALYSTS...81
II.2.1. INTRODUCTION...81
II.2.2. BIBLIOGRAPHY: REDUCTION AND OXIDATION STEPS OF PD...81
II.2.2.1. Effect of Pd on CH4 activation and oxidation ...81
II.2.2.2. Studies of palladium oxidation...82
II.2.2.2.1. Experimental study...82
II.2.2.2.1.1. Pd single crystals and polycrystalline Pd foils ...82
II.2.2.2.1.2. Pd supported catalysts ...83
II.2.2.2.2. Theoretical approach of palladium oxidation...85
II.2.2.2.2.1. Initial oxidation ...85
II.2.2.2.2.2. Theories of metal oxidation: generalities ...86
II.2.2.2.2.3. The Cabrera-Mott theory...86
II.2.2.2.3. From experiment to model ...86
II.2.2.3. Studies of palladium reduction...87
II.2.2.3.1. Reduction by H2 and CH4...87
II.2.2.3.2. Reduction model...88
II.2.2.4. Conclusion...88
II.2.3. EXPERIMENTAL PART...89
II.2.3.1. Sample preparation ...89
II.2.3.1.1. Support ...89
II.2.3.1.2. Pd catalysts ...89
II.2.3.2. Physical characterizations ...89
II.2.3.2.1. X-ray diffraction...89
II.2.3.2.2. Transmission Electron Microscopy...89
II.2.3.3. Chemical characterizations...89
II.2.3.3.1. Atomic absorption ...89
II.2.3.3.2. H2 chemisorption...89
II.2.3.3.3. CO chemisorption...90
II.2.3.3.4. O2-Temperature Programmed Oxidation (TPO) ...90
II.2.3.3.5. CH4-Temperature Programmed Reduction (TPR) ...90
II.2.3.4. Catalytic test...91
II.2.3.4.1. Apparatus...91
II.2.3.4.2. Catalytic steps...91
II.2.4. RESULTS AND DISCUSSION...93
II.2.4.1. Characterization...93
II.2.4.2. Temperature Programmed Combustion (TPC)...94
II.2.4.3. Temperature step conditions ...95
II.2.4.3.1. Reduction steps...95
II.2.4.3.2. Oxidation step...96
II.2.4.3.2.1. Stabilization of the catalysts...96
II.2.4.3.2.2. Reoxidation time of Pd metal...99
II.2.4.3.2.3. Activation energy and turnover frequency...100
II.2.4.3.3. Remarks...101
II.2.4.4. CH4- Temperature Programmed Reduction (TPR)...102
II.2.5. CONCLUSION...103
II.3. APPLICATION TO MONOLITH CATALYSTS...105
II.3.1. INTRODUCTION...105
II.3.2. EXPERIMENTAL PART...105
II.3.2.1. Preparation of the 15wt%(1wt%Pd/Al2O3)/cordierite monolith...105
II.3.2.2. Catalytic test...105
II.3.3. RESULTS AND DISCUSSION...105
II.3.3.1. 15wt%(1wt%Pd/Al2O3)/cordierite monolith ...105
II.3.3.1.1. Catalyst stabilisation after treatment under O2/N2 at 600°C ...105
II.3.3.1.2. CH4 pulse without presence of O2 in the gas feed...106
II.3.3.1.3. C3H6 pulse without presence of O2 in the gas feed ...107
II.3.3.1.4. C3H6 pulse with presence of O2 in the gas feed ...107
II.3.3.1.4.1. C3H6 pulse of 2 min...107
II.3.3.1.4.2. C3H6 pulse of 10 s ...108
II.3.3.1.5. Energetic evaluation ...109
II.3.3.1.5.1. Pd° and CH4 oxidation after the reduction pulse...109
II.3.3.1.5.2. Adiabatic temperature ...109
II.3.3.2. Monolith supplied by Renault SAS ...110
II.3.3.2.1. Catalyst stabilisation after treatment under O2/N2 at 600°C ...110
II.3.3.2.2. Temperature during C3H6 pulse step ...111
II.3.3.2.3. Influence of the C3H6 pulse time...111
II.3.3.2.4. Influence of the presence of C3H6 in the gas feed...112
II.3.3.2.5. Lower temperature ignition of the catalyst...112
II.3.4. CONCLUSION...113
II.4. APPENDIX...114
POISONING...116
II.5. REFERENCES BIBLIOGRAPHIQUES...117
CHAPITRE III : OXYDATION DU METHANE A HAUTE TEMPERATURE – METHANE OXIDATION AT HIGH TEMPERATURE...119
III.1. INTRODUCTION...119
III.2. HIGH CATALYTIC ACTIVITY AND STABILITY OF PD DOPED MN-BASED MATERIALS CATALYSTS FOR THE CH4 CATALYTIC COMBUSTION ...120
III.2.1. RESUME DE L’ARTICLE 1 ...120
III.2.2. ARTICLE 1...120
III.3. EFFECT OF THE PD PRECURSOR SALT ON THE ACTIVITY AND STABILITY OF PD DOPED MN-BASED CATALYSTS FOR THE CH4 CATALYTIC COMBUSTION UNDER SEVERE CONDITIONS...132
III.3.1. RESUME DE L’ARTICLE 2 ...132
III.3.2. ARTICLE 2...132
III.4. EFFECT OF PALLADIUM ON THE REDUCIBILITY OF MN BASED MATERIALS: CORRELATION WITH METHANE OXIDATION ACTIVITY...142
III.4.1. RESUME DE L’ARTICLE 3 ...142
III.4.2. ARTICLE 3...142
Introduction générale
A l’heure actuelle, jamais la géopolitique énergétique n’a été aussi complexe. La constante croissance économique de la Chine et de l’Inde nécessite des besoins de plus en plus importants en énergie primaire (pétrole, gaz, charbon). Ces pays représentent à eux seuls environ 30 % de la consommation totale en énergie dans le monde (12 GTep.an-1 [a]). Cela implique une pression grandissante sur les ressources (menaces d’approvisionnements) sans compter sur les incidents climatiques impliqués par les problématiques de production et de transports (Golfe du Mexique, fuite d’un oléoduc en Alaska). Les échanges mondiaux sont aussi soumis à des tensions plus marquées (entre la Russie et l’Europe pour le gaz, au Moyen-Orient pour le pétrole). Afin de prendre en compte l’hypothèse d’une éventuelle pénurie des matières premières énergétiques non renouvelables (pétrole, gaz, charbon), qui impliquerait la fin d’une énergie abondante et peu chère, le mot d’ordre actuel du point de vue économique est l’efficacité énergétique, c’est à dire, à service équivalent, consommer moins d’énergie. D’un point de vue environnemental, l’augmentation de la consommation d’énergie se traduit par des émissions de produits polluants à tous les niveaux (local, régional et global) [1].
Les travaux de recherche de cette thèse, réalisés en partenariat avec l’ADEME (Agence De l’Environnement et de la Maîtrise de l’Energie) et cofinancés par Renault SAS, ont pour objectif la réduction des gaz polluants (teneurs et/ou degré de toxicité) provenant d’une source consommatrice d’énergie fossile, le transport routier avec le traitement des gaz d’échappement issus des véhicules particuliers et plus précisément des moteurs de type Diesel. Avant même de présenter avec plus de précision les objectifs de la thèse, un bref récapitulatif sur l’évolution des normes sur les gaz à effet de serre et sur la problématique environnementale liée au transport sera présenté.
L’origine réglementaire des émissions de gaz à effet de serre provient d’un constat alarmant sur le réchauffement climatique. L'effet de serre est certes un phénomène naturel, sans lequel la température moyenne à la surface de la Terre serait d'environ − 18 °C, mais celui-ci est intensifié par les activités humaines qui produisent de grandes quantités de dioxyde de carbone (CO2), d'oxydes
d'azote (NOx), de méthane (CH4), d'ozone troposphérique (O3) et de chlorofluorocarbones (CFC)
[2]. La signature de la convention sur les changements climatiques lors de la Conférence de Rio de Janeiro du 3 au 14 juin 1992 marque les prémisses d’une tentative de coopération internationale. Cette convention ne concerne en réalité que le réchauffement du climat mondial. Son objectif est de « stabiliser, conformément aux dispositions pertinentes de la Convention, les concentrations de gaz à effet de serre dans l'atmosphère à un niveau qui empêche toute perturbation anthropique dangereuse du système climatique » (article 2). Par la suite, des avancées ont eu lieu, avec l'arrêté du Protocole de Kyoto le 15 mars 1999 qui marque le début d’un réel effort planétaire. Protocole complexe de par la juxtaposition de tendances opposées, ses principales caractéristiques sont : (i) l'énumération des gaz à effet de serre, (ii) l'acceptation par des pays industrialisés d'objectifs de réduction des émissions sans contrepartie de la part des pays en développement, (iii) la reconnaissance du rôle des puits et des réservoirs de gaz à effet de serre et leur inclusion dans les objectifs et (iv) la possibilité de totaliser les émissions des pays industrialisés lorsqu'il s'agit de réduire les émissions et la possibilité de céder des « droits d'émissions » entre États ou entre un État et une personne privée. Le Protocole parle « d'un mécanisme propre qui devra être défini ». La mise en œuvre du Protocole a révélé la bonne attitude de l'Europe : entre 1990 et 2005, la Communauté européenne a réduit ses émissions de CO2 d'environ 1,5 % alors que l'ensemble des autres États
membres de l'OCDE les a augmenté de 8 %. En particulier, l'Australie, le Canada et les États-Unis ont augmenté chacun leurs émissions de 7 à 9 %. Le 25 juillet 2001, les 180 membres de la convention des Nations Unies sur les changements climatiques, réunis à Bonn, sont parvenus à un accord politique sur les modalités d'application du Protocole de Kyoto. Les Accords de Marrakech du 10 novembre 2001, ont fixé les règles de mise en œuvre du Protocole de Kyoto. Les ministres de l'Environnement des États membres de l'Union européenne ont approuvé la ratification du Protocole de Kyoto le 4 mars 2002, ouvrant la voie à une ratification par chaque État membre. Rappelons que pour entrer en vigueur, le Protocole de Kyoto doit être ratifié par au moins 55 États, représentants au moins 55 % des rejets de CO2 en 1990. Les États-Unis ayant rejeté le traité, l'entrée en vigueur
du Protocole repose actuellement sur sa ratification par la Russie.
Le secteur du transport routier a une part importante dans l’émission des gaz à effet de serre ainsi que dans l’augmentation de la consommation d’énergie. En vu de remplacer les carburants d’origine fossile, des solutions techniques existent mais leurs impacts restent à évaluer. L’utilisation de biocarburants de première génération est sujet à caution avec les problèmes actuels sur les denrées alimentaires. Au contraire, les carburants de seconde génération, utilisant la biomasse lignocellulosique, n’entrent pas en concurrence avec les matières premières alimentaires. Les nouvelles technologies telles que les piles à combustibles avec l’emploi ou non d’un reformeur interne posent le problème du combustible (production d’hydrogène ou d’éthanol). Donc, malgré une substitution grandissante mais limitée des carburants à base de pétrole, majoritairement et de charbon (carburants de synthèse), le pétrole reste et restera pour les années à venir la principale source de production de carburants. Les moteurs à allumage commandé et Diesel, utilisant respectivement l’essence et le gazole comme carburant, présentent des caractéristiques techniques différentes mais la nature de leurs émissions polluantes en plus du CO2 est identique. Les
principaux polluants des moteurs essence sont le CO, les NOx et les hydrocarbures (HC) imbrûlés. Pour le moteur diesel, en plus de ces polluants s’ajoute la présence de suie à traiter. Les suies sont présentes en essence mais leur nombre et leur taille sont plus faibles. Avant même la mise en place du protocole de Kyoto, en Europe, les gouvernements en accord avec les industriels automobiles ont instauré des normes sur les émissions de polluants (normes EURO) pour les véhicules particuliers [3, 4]. Les différentes teneurs en polluant admises pour chaque type de moteur sont résumées dans le Tableau 1. Afin d’éliminer ces polluants, différentes technologies sont appliquées suivant le type de motorisation. Le Tableau 2 présente ces systèmes de dépollution. Les émissions des véhicules sont ensuite évaluées lors de cycles standardisés, cycles urbains (UDC : Urban Driving Cycle) et extra-urbains (EUDC : Extra Urban Driving Cycle). La quantité équivalente de CO2 émis a permis
la mise en place obligatoire depuis le 10 mai 2006 (arrêté publié au J.O. le 10 novembre 2005) d'une étiquette-énergie sur les ventes de voitures neuves. La note attribuée évolue en fonction du taux d’émissions de CO2, “A” pour les véhicules émettant une teneur inférieure à 100 gCO2.km-1 à
“F” pour les véhicules les plus pollueurs, teneur supérieure à 250 gCO2.km-1 [5].
Tableau 1: Normes européennes pour les véhicules particuliers (E : essence ; D : Diesel ; * : HC+NOx).
Polluants (g.km-1)
Normes Années Moteur
CO HC NOx Particules E 2,72 --- --- EURO I 1993 D 2,72 0,97* 0,14 E 2,20 --- --- EURO II 1996 D 1,00 0,71* 0,08 E 2,20 0,20 0,15 --- EURO III 2000 D 0,64 --- 0,50 0,05 E 1,00 0,10 0,08 --- EURO IV 2005 D 0,50 0,30* 0,25 0,025 E 1,0 0,10 0,06 0,005 EURO V 2009 D 0,50 0,23* 0,18 0,005
Introduction générale Tableau 2: Technologies d'élimination des émissions polluantes en fonction du type de motorisation.
Technologies Fonctions Moteur
Catalyseur « trois voies » Oxydation du CO et des HC + Réduction des NOx Essence
Catalyseur d’oxydation Oxydation du CO et des HC Diesel
De-NOx Réduction sélective catalytique (SCR)
Piège à NOx (NOx-trap) Diesel
Filtre à particules Particules Diesel
L’évolution vers des normes de plus en plus sévères en matière de dépollution automobile pousse les industriels à se poser la question du traitement du méthane contenu dans les gaz d’échappement des moteurs Diesels. En effet, les normes imposées en Europe prennent en compte les hydrocarbures imbrûlés de façon globale, incluant le méthane. Le pouvoir de réchauffement global du méthane étant 22 fois celui du CO2 (CO2 étant la référence à 1) avec un temps de séjour de 12
ans (200 ans pour le CO2), la question du traitement du méthane (molécule très stable et très
difficile à oxyder avec les systèmes actuels du fait de sa symétrie et de la force des liaisons C-H) devient donc centrale pour l’homologation des futurs véhicules si les constructeurs veulent diminuer leur niveau d’émission en HC. Cette question se pose d’autant plus que l’impact du méthane sur l’effet de serre est non négligeable. Dans ces conditions, nous pouvons même nous poser la question de l’apparition d’une norme spécifique sur les émissions de méthane, d’autant que nous assistons à une augmentation prévisible de la teneur en méthane dans les gaz d’échappement. Cette augmentation s’explique par les orientations technologiques suivantes de la part des constructeurs automobiles. Une première “source” de méthane est la purge du piège à NOx ou NOxtrap. Le piège à NOx est la solution technologique retenue par les constructeurs automobiles sur les véhicules particuliers pour atteindre le niveau requis par EuroVI. Dans son fonctionnement, le NOx-trap nécessite des passages en richesse supérieure à 1 pour la désorption et la réduction des NOx stockés. Ce mode de fonctionnement est réalisé dans le moteur par injection retardée : les hydrocarbures sont injectés au moment de la détente des gaz, ainsi ils sont vaporisés dans le cylindre pour former un gaz riche en réducteurs à l’échappement (richesse voisine de 1,05). Pour indication, la quantité de méthane émis à l’échappement lors des purges d’un NOx-trap peut atteindre 30 % du niveau de la norme EuroV sur les HC. Une deuxième “source” est le mode de combustion homogène HCCI (Homogeneous Charge Compression Ignition). Ce type de motorisation est à l’étude car très prometteur entre autres pour la réduction des émissions de NOx et de particules. Ce moteur fonctionne avec un mélange pauvre de richesse supérieure à celui d'un moteur Diesel conventionnel. La proportion de méthane dans les hydrocarbures imbrûlés peut atteindre 10 % en masse et constituer jusqu’à deux fois le niveau de norme EuroV en HC.
Le premier objectif de la thèse est donc de développer un système permettant l’oxydation à basse
température du méthane (250 °C). Les solutions conventionnelles mises en place sur véhicules particuliers, tel le catalyseur d’oxydation, présentent des efficacités de conversion très faibles du méthane. En effet, l’amorçage de l’oxydation du méthane (T50) ne se produit qu’après 450 °C [b],
température rarement atteinte en Diesel au cours d’un cycle d’homologation. Pour cette raison, Renault SAS a donc décidé de s’orienter vers un procédé innovant, le procédé plasma/catalyse. En effet, la technique plasma semble être une alternative aux techniques de catalyse conventionnelles spécialement pour des concentrations très faibles en hydrocarbures (< 1 g.m-3) [6] et elle permet aussi d’abaisser les températures d’activation actuelles. La température recherchée pour l’activation
de cette réaction est la température moyenne rencontrée sur cycle à l’échappement du moteur Diesel, de l’ordre de 250°C.
Après avoir étudié, comparé et réalisé un bilan sur les limites des différents procédés plasmas, nous avons focalisé nos recherches sur la catalyse hétérogène (hors plasma) avec la compréhension globale des mécanismes qui entrent en jeu lors de l’oxydation du méthane (étude de la phase active, relation avec le support, …) à basse et haute température sur des matériaux de type Pd/Al2O3 et d’en
améliorer la composition.
Les catalyseurs Pd/Al2O3 ont été largement étudiés [7, 8]. A basse température, ce type de
matériaux possèdent une très bonne activité catalytique envers le méthane. Il est généralement admis que le palladium sous sa forme oxyde (PdO) est le métal noble le plus actif pour l’oxydation du méthane en milieu pauvre (excès d’oxygène). Cependant, l’alternance de traitements oxydants et réducteurs a montré un gain de conversion du méthane. Le deuxième objectif, réalisé au sein de l’équipe du Professeur Pio Fortazzi du Politecnico di Milano en 2007, est de comprendre les paramètres influençant l’oxydation du palladium métallique (Pd°) et la réduction de la phase oxyde (PdO) déposées sur un support alumine pour la réaction d’oxydation du méthane (mélange réactionnel synthétique : O2/CH4/H2O/He) à basse température (T < 500 °C). L’étape limitante pour
l’oxydation du méthane étant l’adsorption et la dissociation de cette molécule à la surface du métal noble [9]. Cependant, à haute température, le catalyseur Pd/Al2O3 présente un désavantage
significatif, une transformation de la phase PdO en Pd° moins actif, entraînant une perte d’activité.
Le troisième objectif est donc de développer de nouveaux matériaux présentant à la fois une bonne
activité catalytique ainsi qu’une très bonne stabilité thermique pour l’oxydation du méthane à des hautes températures, par exemple > 500 °C, températures éventuellement rencontrées lors de la phase de purge du système NOx-trap. Les matériaux à structure hexaaluminate (formule générale BaAl12O19) sont de bons candidats pour la substitution du système Pd/Al2O3 car ils présentent une
excellente stabilité thermique. Ces travaux portent, d’une part, sur le développement d’une nouvelle voie de préparation des matériaux haxaaluminates dopés (La, Sr, Mn) afin de trouver le meilleur compromis entre activité catalytique et stabilité thermique. D’autre part, l’influence de la nature du sel précurseur du métal noble Pd (nitrate ou organique) sur le gain d’activité a été étudiée. Enfin, des recherches ont été menées sur la relation entre le gain d’activité catalytique et de stabilité thermique et l’interaction du couple PdO/Pd° et du couple redox Mn3+/Mn2+.
La rédaction du manuscrit s’organise en trois chapitres, suivant les trois objectifs présentés ci-dessus. Le premier chapitre est dédié à la faisabilité des différents systèmes plasma/catalyse pour abattre le méthane à des températures inférieures à 250 °C. Le second chapitre présente les résultats obtenus sur la compréhension des mécanismes d’activation du catalyseur Pd/Al2O3 pour l’oxydation
du méthane à basse température. Enfin, le troisième chapitre, recueil des articles publiés, rassemble les résultats obtenus sur les matériaux développés pour l’oxydation catalytique du méthane pour palier au problème de stabilité thermique du système Pd/Al2O3 à haute température. Afin de suivre
le déroulement de la thèse, un volet en fin de manuscrit pouvant servir de marque-page, présente la structure du document.
Références bibliographiques
Références bibliographiques
[1] J.P. Favennec, Géopolitique de l’énergie, IFP Publications, Editions TECHNIP 2007. [2] GIEC, Groupe Intergouvernemental sur l’Etude du Climat.
http://ipcc-wg1.ucar.edu/wg1/wg1-report.html [24/04/2008] [3] Rhodia. Catalysis. Dépollution automobile.
http://www.rhodia-ec.com/site_ec_fr/catalyse/index_depollution.htm [22/11/2005] [4] Diesel. Emission standards.http://www.dieselnet.com/standards/#eu [22/11/2005] [5] Ademe. http://www2.ademe.fr/ [20/01/2008]
[6] F. Holzer, U. Roland, F.D. Kopinke, Appl. Catal. B 38 (2002) 163-181. [7] T. V. Choudhary, S. Banerjee, V. R. Choudhary, Appl. Catal. A 234 (2002) 1.
[8] K. Persson, A. Ersson, S. Colussi, A. Trovarelli, S. G. Jaras, Appl. Catal. B 66 (2006) 175. [9] D. Ciuparu, M.R. Lyubovsky, E. Altamn, L. Pfefferle, A. Datye, Catal. Rew. 44 (2002) 593.
I.1. PARTIE BIBLIOGRAPHIE ...6
I.1.1. LE PLASMA...6
I.1.2. PLASMA NON-THERMIQUE...6
I.1.3. PLASMA THERMIQUE...8
I.1.4. CONFIGURATIONS TECHNOLOGIQUES...11
I.1.4.1. Configurations de décharges...11
I.1.4.2. Configuration plasma/catalyseur ...11
I.1.5. REACTION DU METHANE DANS UN PLASMA D’AIR...12
I.1.6. RESULTATS EXPERIMENTAUX LIES A L’OXYDATION DU METHANE...12
I.1.6.1. Influence du plasma sur le CH4 seul...12
I.1.6.2. Influence du plasma sur un mélange CH4/O2...13
I.1.6.3. Influence du plasma sur un mélange CH4/H2O ...14
I.1.6.4. Influence du plasma sur un mélange CH4/CO2...15
I.1.6.5. Influence du plasma sur un mélange CH4/O2/H2O...16
I.1.6.6. Influence du plasma sur un mélange CH4/O2/CO2/NO ...16
I.1.6.7. Remarque sur la formation d’ozone ...17
I.1.7. CONCLUSION...17
I.2. PARTIE EXPERIMENTALE GENERALE ...20
I.2.1. MONTAGE D’OXYDATION...20
I.2.2. ANALYSES CHIMIQUES ET PHYSICO-CHIMIQUES...20
I.2.2.1. Diffraction des rayons X...20
I.2.2.2. Analyses des produits de réaction ...20
I.2.3. MESURE DES COMPOSANTES ELECTRIQUES : TENSION U(T) ET INTENSITE I(T) ...21
I.2.3.1. Oscilloscope numérique...21
I.2.3.2. Sonde haute tension et sonde d’induction ...21
I.2.4. CALCUL DE PUISSANCE...22
I.2.4.1. Alimentation continue...22
I.2.4.2. Alimentation sinusoïdale ...22
I.2.4.2.1. La méthode du condensateur de Manley ...22
I.2.4.2.2. Les phases d’un plasma ...24
I.2.4.3. Alimentation impulsionnelle ...24
I.3. PLASMA NON-THERMIQUE : RESULTATS ET DISCUSSIONS...25
I.3.1. CONFIGURATION DBD : REACTEUR COAXIAL, ALIMENTATION IMPULSIONNELLE...25
I.3.1.1. Introduction ...25
I.3.1.2. Partie expérimentale...25
I.3.1.2.1. Schéma du montage...25
I.3.1.2.2. Préparation du catalyseur LACCO ...26
I.3.1.3. Effet du plasma seul...26
I.3.1.4. Etude de l’interaction plasma/catalyse ...29
I.3.1.4.1. Remarque : modification du réacteur ...29
I.3.1.4.2. Influence de la position du catalyseur (POST ou IN-plasma) ...30
I.3.1.4.3. Température d’activation du catalyseur...33
I.3.1.5. Conclusion ...34
I.3.2. CONFIGURATION DBD : REACTEUR COAXIAL, ALIMENTATION SINUSOÏDALE...35
I.3.2.1. Introduction ...35
I.3.2.2. Partie expérimentale...35
I.3.2.2.1. Schéma du montage et caractéristiques des réacteurs ...35
I.3.2.3. Réacteur quartz...36
I.3.2.3.1. Comportement sous mélange N2/O2...36
I.3.2.3.2. Oxydation du CH4...37
I.3.2.3.3. Influence de H2O, du CO2 et du volume plasma ...38
I.3.2.3.3.1. Influence de H2O et du CO2...38
I.3.2.3.3.2. Influence du volume plasma...38
I.3.2.3.4. Comparaison entre alimentation impulsionnelle et alimentation sinusoïdale ...39
I.3.2.3.5. Présence d’un solide IN-plasma ...40
I.3.2.3.5.1. Effet de billes de Al2O3 imprégnées ou non à 1%Pd...40
I.3.2.3.5.2. Effet d’un matériau ferroélectrique BaTiO3...40
I.3.2.4. Réacteur céramique ...46
I.3.2.4.1. Phénomène parasite dû à la conception du réacteur ...47
I.3.2.4.2. Oxydation du CH4 après modification du réacteur...47
I.3.2.4.3. Conditions expérimentales...47
I.3.2.4.5. Résultats...48
I.3.2.4.6. Remarques ...50
I.3.2.5. Conclusion ...51
I.3.3. CONFIGURATION DBD : REACTEUR PLAN/PLAN, ALIMENTATION SINUSOÏDALE...52
I.3.3.1. Introduction ...52
I.3.3.2. Partie expérimentale...52
I.3.3.3. Résultats...52
I.3.3.4. Remarques ...53
I.3.3.5. Conclusion ...54
I.3.4. CONFIGURATION POINTE/PLAN : ALIMENTATION CONTINUE...55
I.3.4.1. Introduction ...55
I.3.4.2. Partie expérimentale...55
I.3.4.3. Résultats et conclusion ...55
I.3.5. CONFIGURATION MULTIPOINTES/PLAN : ALIMENTATION IMPULSIONNELLE...56
I.3.5.1. Introduction ...56
I.3.5.2. Partie expérimentale...56
I.3.5.3. Résultats...56
I.3.5.4. Conclusion ...57
I.4. PLASMA THERMIQUE : RESULTATS ET DISCUSSIONS...58
I.4.1. CONFIGURATION POINTE/PLAN : ALIMENTATION SINUSOÏDALE...58
I.4.1.1. Introduction ...58
I.4.1.2. Partie expérimentale...58
I.4.1.3. Résultats...60
I.4.1.3.1. Etude énergétique ...60
I.4.1.3.1.1. Détermination de la chaleur de combustion ...60
I.4.1.3.1.2. Température adiabatique ...60
I.4.1.3.2. Influence de la composition du mélange gazeux...61
I.4.1.3.2.1. Variation de la concentration initiale en CH4...61
I.4.1.3.2.2. Présence de C3H6 dans le mélange gazeux ...62
I.4.1.3.3. Influence du gap entre les électrodes...63
I.4.1.3.3.1. Conversion du CH4...63
I.4.1.3.3.2. Mesure de température ...64
I.4.1.3.4. Sens de circulation du gaz par rapport à l’arc électrique...66
I.4.1.3.5. Utilisation d’un catalyseur en position POST-plasma...66
I.4.1.3.5.1.Traitement du CH4 seul ...66
I.4.1.3.5.1. Traitement d’un mélange (CH4+C3H6)...67
I.4.1.3.7.1. Paramètres électriques ...68
I.4.1.3.7.2. Produits de réaction : O3, CO et NOx...69
I.4.1.3.8. Conclusion ...69
I.4.2. CONFIGURATION POINTE/PLAN : ALIMENTATION IMPULSIONNELLE...71
I.4.2.1. Introduction ...71
I.4.2.2. Partie expérimentale...71
I.4.2.2.1. Générateur impulsionnel...71
I.4.2.2.2. Montage plasma...71
I.4.2.3. Résultats...72
I.4.2.3.1. Puissance consommée par le plasma ...72
I.4.2.3.2. Conversion du méthane ...73
I.4.2.4. Conclusion ...74
I.5. CONCLUSION: COMPARAISON DES DIFFERENTES SYSTEMES PLASMA/CATALYSE...75
I.1. Partie bibliographie
Chapitre I : Technologie plasma/catalyse
I.1. Partie bibliographie
I.1.1. Le plasma
Dans la nature, les plasmas constituent le quatrième état de la matière après les états solide, liquide et gazeux. Le terme plasma désigne un gaz partiellement ionisé qui peut être produit par une décharge électrique, et est caractérisé par le comportement des particules chargées. Le plasma, considéré comme un système de conversion d’énergie, transforme l’énergie électrique en énergie lumineuse, cinétique ou chimique. C’est cette propriété de source d’espèces chimiquement actives qui est recherchée dans les applications de traitement de surfaces ou de procédés en volume telles que le traitement des matériaux ou la dépollution de l’air.
La science du plasma regroupe différents types de plasma, les plasmas à basse (< 100 mBar) ou haute pression (> 100 mBar), les plasmas micro-onde, radio fréquence, basse fréquence, les plasmas thermiques, les plasmas très fortement hors équilibre à la pression atmosphérique ou les plasmas froids. Dans la suite sont décrits les plasmas non-thermique et thermique à la pression atmosphérique.
I.1.2. Plasma non-thermique
La technologie plasma froid est la plus répandue pour le traitement des gaz. Il est à noter que dans la littérature, ce type de plasma peut être désigné de différentes manières :
- Plasma Basse Température (Low Temperature Plasma) - Plasma Luminescent (Glow Plasma)
- Décharge à Barrière Diélectrique (Dielectric Barrier Discharge) - Plasma Non-Thermique (Non-Thermal Plasma)
- Plasma Hors Equilibre (Non Equilibrium Plasma)
- Plasma Froid Atmosphérique (Cold Atmospheric Plasma)
Le plasma froid correspond à un plasma faiblement ionisé dont les propriétés sont dominées par les collisions des particules chargées avec les atomes ou molécules neutres majoritaires. Dans un plasma hors équilibre, les différentes particules ont donc des énergies moyennes très différentes. Par exemple, l’énergie moyenne des électrons peut-être de plusieurs eV (1 eV ~ 11600 K), tandis que la température du gaz ne dépasse pas quelques centaines de degrés Kelvin. Si la puissance dissipée par unité de volume devient trop élevée, le plasma passe d’un système hors équilibre à un système proche de l’équilibre thermodynamique (plasma thermique).
Pour obtenir un plasma froid, les électrons sont accélérés par un fort champ électrique et transmettent leur énergie aux espèces neutres sous forme d’excitation, ionisation, collisions inélastiques. Les processus réactionnels mis en jeu dans le plasma et les énergies nécessaires à la rupture des liaisons de quelques composés sont présentés sur la Figure 1 et dans le Tableau 1 [1-2]. La densité d’énergie fournie est suffisamment faible pour que les atomes et molécules excités ne restituent que partiellement cette énergie aux électrons. Le bilan énergétique est déséquilibré, et ce sont donc principalement les électrons, qui sont accélérés par le champ électrique, qui transmettent l’énergie dans le milieu. La température électronique est donc grande devant celle des ions et des espèces non chargées, ce qui permet de créer un milieu chimiquement actif à basse température. L’obtention de ce milieu réactionnel est facile de mise en œuvre.
Radicaux 1aires OH, O, N radical / radical radical / neutre Temps (s) 10-14 10-12 10-10 10-8 10-6 10-4 10-2 100
Processus primaire Processus secondaire
Radicaux Transfert de charges A+ + B → A + B+ Ionisation A + e-→ A* + e- + e -Excitation A + e-→ A* + e -Désexcitation Transfert d’énergie Propagation du streamer Hυ ion / ion Radicaux 2aires HO2, O3 Recombinaisons
Figure 1: Evolution temporelle de la formation de la décharge. Tableau 1: Energies de dissociation des liaisons de certains composés [2].
Composés Liaison Energie (eV) Composés Liaison Energie (eV)
CH4 C-H 4,47 O2 O-O 5,11 CH3CH3 C-H 4,29 CO C-O 11,10 CH3CH3 C-C 3,80 CO2 C-O 5,45 CH2=CH2 C-H 4,33 H2O O-H 5,11 CH2=CH2 C-C 7,45 HO* O-H 4,40 N2 N-N 9,75
Le plasma froid peut être généré soit par les techniques à faisceaux d’électrons soit par les techniques à décharges. C’est cette dernière qui est utilisée ici. Les électrons sont directement créés et accélérés, à pression atmosphérique, dans le volume du gaz à traiter par l’application d’une haute tension entre deux électrodes. Les électrons collisionent avec les molécules du gaz vecteur et transfèrent une partie de leur énergie à mesure qu’ils circulent d’une électrode vers l’autre.
Les différentes phases du plasma sont les suivantes :
- Formation de la décharge : l’application d’une haute tension entre deux électrodes induit l’apparition d’un champ électrique E à l’intérieur du gaz contenu entre les électrodes. Le champ réduit (Ered) est défini comme le rapport entre E, le champ électrique induit et N, la
densité de molécules neutres du gaz : Ered(V.cm2) = E(V.cm-1)/N (cm-3). A pression
atmosphérique, la décharge se manifeste par une multitude de filaments ou de canaux. La formation de ces canaux et donc de la décharge s’explique par la théorie des streamers et se déroule en deux phases : la phase d’avalanche et la phase de streamer.
- La phase d’avalanche : après application d’une tension, les électrons acquièrent une certaine énergie cinétique. Sur leur trajet, ils vont rentrer en collision avec les molécules du gaz et perdre une partie de leur énergie. Si l’énergie de l’électron est suffisante, en l’occurrence supérieure au potentiel d’ionisation de la molécule, celle-ci est alors ionisée par collision électronique entraînant la libération d’un électron. L’électron projectile et l’électron fraîchement libéré sont accélérés par le champ électrique et vont induire d’autres réactions d’ionisation libérant d’autres électrons. C’est le phénomène d’avalanche électronique.
I.1. Partie bibliographie
- La phase streamer : A haute pression (> 100 mBar), après la formation de l’avalanche, la différence de mobilité entre électrons et ions va induire une charge d’espace qui grandit au fur et à mesure du développement de l’avalanche. Lorsque le nombre d’électrons devient de l’ordre de 108 à 109, le champ positif de charge d’espace associé devient plus important que le champ Laplacien et gouverne alors la dynamique de décharge. L’onde d’ionisation se propage sous l’effet de son propre champ de charge d’espace. C’est cette onde qui est appelée streamer [3]. Une fois le stremaer propagé une microdécharge est établie et l'émission des états excités de l'azote est observée (Figure 2). Le courant intense qui circule par ces canaux favorise d’autres processus au sein du gaz en plus de l’ionisation : excitation (M + e- → M* + e-), désexcitation (M* → M + hυ), photo-ionisation (M + hυ → M+ + e-) et
recombinaison (M+ + e-→ M + hυ). Par photo-ionisation, de nouveaux électrons sont formés qui sont accélérés par le champ E régnant dans le streamer. De nouvelles réactions sont induites et le processus se répète faisant progresser le streamer. Ces nombreuses réactions induisent une chimie très complexe.
Figure 2: Multiples filaments de plasma obtenus dans l'air à pression atmosphérique pour une série de décharges créées entre un fil et une électrode plane [a].
Ces différentes phases peuvent être générées par une alimentation électrique sinusoïdale, continue ou monopolaire. Un exemple de signal de sortie d’une alimentation monopolaire est présenté sur la Figure 3. Lors d’une impulsion, le phénomène se décompose en plusieurs phases:
- Phase 1 : génération de l’impulsion positive, les électrodes se chargent.
- Phase 2 : phase d’attente, tous les interrupteurs du système sont ouverts. Durée 0,5-1 μs. - Phase 3 : phase de roue libre, l’énergie stockée par les électrodes est déchargée. Ainsi le cycle suivant peut démarrer sans énergie résiduelle. Durée 80 μs.
Tension sur la charge
Courant dans la charge
+HT
Tp=0,5-1μs Td=1μs
1 2 3
Tension sur la charge
Courant dans la charge
+HT
Tp=0,5-1μs Td=1μs
1 2 3
Figure 3 : Signal de sortie d’une alimentation monopolaire (échelle de temps non uniforme).
I.1.3. Plasma thermique
Dans le cas présent, le plasma thermique est établit sous la forme d'un arc électrique fonctionnant à une pression voisine de la pression atmosphérique. Ce plasma est un gaz partiellement ou totalement ionisé dans lequel toutes les particules ont à peu près la même énergie cinétique moyenne. Cette décharge est caractérisée par une intensité élevée (kA), un champ électrique faible
(V.cm-1) et donc une tension faible et sensiblement proportionnelle à la longueur de l’arc [3]. Dans le cas d'un isolant gazeux, la rigidité diélectrique (valeur maximum du champ électrique que le milieu peut supporter avant le déclenchement d’un arc électrique) dépend de la pression du gaz, selon la Loi de Paschen. Cette loi indique que l'apparition d'un arc électrique dans un gaz, pour une certaine valeur du champ électrique v de claquage (dit champ disruptif), est une fonction généralement non linéaire du produit de la pression P du gaz par la distance d entre les électrodes divisé par la température T : v=f(P×d/T). Il existe toujours une tension électrique minimale pour une certaine distance entre les électrodes (champ disruptif minimal, qui est une tension électrique par unité de longueur s'exprimant dans ce cas classiquement en kV.mm-1) à une pression donnée, permettant au courant électrique de circuler dans l’espace inter-électrodes : cette valeur est appelée le minimum de Paschen. Le champ disruptif de l'air sec est évalué à 3600 V.mm-1 à la pression atmosphérique. Il peut descendre à un seuil de 1000 V.mm-1 dans un air saturé en humidité, ceci pour des distances inter-électrodes proches (de l'ordre du millimètre ou du centimètre). Pour des distances plus grandes, ce champ disruptif est encore plus élevé (Figure 4) car il n'y a pas assez d'électrons libres et leur libre parcours moyen est trop faible pour qu'ils puissent être accélérés suffisamment entre deux collisions (leur énergie cinétique est insuffisante pour ioniser le gaz). Cependant, à distance inter-électrodes identique, plus la pression de l'air diminue et plus la décharge électrique survient à des tensions faibles. Cela se traduit par une valeur minimale sur la courbe de Paschen. Par contre, si la pression continue de descendre sous ce minimum alors la tension à fournir augmente à nouveau (la courbe remonte). Le libre parcours moyen des électrons devient cette fois trop grand : il n'y a plus assez d'atomes sur leur chemin pour déclencher, par collisions avec ceux-ci, le phénomène d'avalanche électronique qui transforme le gaz en plasma.
Figure 4: Influence de la distance entre les électrodes sur la tension de claquage [4].
La position d'un arc électrique repose sur le principe de l'énergie minimale, c’est à dire qu’il choisit la distance la plus courte et il y reste. Un courant traversant un arc électrique est généralement intense et variable. L’ensemble du montage électrique générant l’arc cause de fortes perturbations électromagnétiques et c’est pourquoi un capteur électrique peut difficilement trouver sa place en sa proximité. Dans le cas d’une application automobile, l’utilisation d’écrans électromagnétiques peut permettre de s’affranchir de ce problème de perturbations. L'écoulement du courant dans la matière ionisée émet un rayonnement de lumière dont le spectre est caractéristique de la nature du gaz, et à un degré moindre, de celle des électrodes dans le cas où ces dernières sont fusibles. Dans le cas général, les arcs émettent une grande proportion d'ultraviolets particulièrement agressifs pour les yeux. Cette ionisation et l'écoulement d'un courant électrique qui s'ensuit engendrent des bruits dus à l'expansion brutale du gaz suite à son échauffement tout aussi brutal.
I.1. Partie bibliographie
Pour qu'il y ait équilibre thermodynamique complet (ETC) avec une seule température relative aux particules : mouvements de translation (collisions élastiques), excitation et ionisation par collisions inélastiques, émission, absorption des photons, il faut que le principe de micro-réversibilité soit satisfait. Ce dernier postule que l'équilibre est maintenu seulement si chaque processus d'interaction est contrebalancé par le même processus inverse. Par exemple, l'émission d'un photon doit être équilibrée dynamiquement par la réabsorption d'un photon. Dans ces conditions, les équations de Maxwell, de Boltzmann, les lois d'action de masse (équilibre chimique) et la loi de Planck (rayonnement du corps noir) sont satisfaites pour une température unique. Tout cela correspond à un plasma contenu dans une cavité hypothétique dont les parois seraient à la température du plasma ou à un plasma dont le volume est suffisamment grand pour que les pertes en périphérie n'interfèrent pas avec l'équilibre au sein du volume. Mais, en général, dans les plasmas de laboratoire, le libre parcours moyen des photons est très grand devant les dimensions du plasma. Cela signifie que les photons émis ne sont pas réabsorbés (ou très peu) et que la loi de Planck n'est pas satisfaite. Le plasma n'est pas un corps noir mais il est totalement ou partiellement optiquement mince. C'est pourquoi le concept d'équilibre thermodynamique local (ETL) a été défini. Il postule que le plasma est optiquement mince (ou partiellement) et que ce sont les processus collisionnels entre particules (et non les processus radiatifs) qui gouvernent les transitions et les réactions au sein du plasma. De plus, cette notion implique que les gradients locaux des propriétés du plasma (température, concentration, ....) soient suffisamment faibles pour que les phénomènes diffusionnels aient le temps de s'équilibrer. Dans ces conditions, le concept d'ETL est valable sauf pour l'équation de Planck. La Figure 5 présente l’évolution de la température du gaz (Tg, particules lourdes) et des électrons (Te) en fonction de la pression dans le plasma [5].
Figure 5: Évolution des températures des électrons Te et des particules lourdes Tg avec la pression dans un plasma d'arc (décharge à courant continu) [5].
Remarque : différenciation entre arc et streamer
Un arc et un streamer sont tous les deux des phénomènes de décharges. Ils créent un canal ionisé où la décharge va pouvoir prendre place. La différence entre un streamer et un arc est que le courant transporté dans un arc est beaucoup plus élevé (de quelques A à quelques dizaines de kA) que dans un streamer [6]. La Figure 6 schématise l’évolution du courant et de la tension lors de la transition entre streamers et arcs. Quand l’arc apparaît, la tension chute et l’intensité augmente brusquement. Les matériaux électrodes sont donc soumis à des échauffements fonction de l'intensité du courant qui les parcourt et doivent donc être dimensionnés afin d’éliminer les calories et d’éviter la fusion de l’électrode métallique.
u
i
Temps
ARC STREAMER
Figure 6: Evolution de u(t) et i(t) lors de la transition entre streamer et arc électrique (sans diélectrique).
I.1.4. Configurations technologiques
Cette partie présente les différentes configurations entre électrodes, diélectrique, plasma et catalyseur, dans le cas du plasma froid et sont largement décrites dans [7]. L’étude du plasma thermique pour le traitement des gaz a été essentiellement limitée à l’utilisation de torches micro-onde. Ces différentes possibilités de mise en œuvre du plasma peuvent être utilisées soit dans le cadre de la synthèse de catalyseurs, soit pour l’activation du catalyseur ou enfin, comme décrit dans la suite, directement pour l’oxydation des hydrocarbures ou des composés organiques volatils (COV). Ces configurations sont décrites ici car elles feront l’objet de renvois dans la suite.
I.1.4.1. Configurations de décharges
La Figure 7 montre trois configurations typiques de décharges [8]. La configuration plan/plan (Figure 7a et Figure 7b) est utilisée pour le traitement de surface. La Figure 7b présente l’avantage d’éviter tout contact entre le plasma et les électrodes métalliques, condition parfois utile lors de l’utilisation de plasmas corrosifs. La configuration coaxiale (Figure 7c) est elle généralement utilisée pour traiter des gaz, que ce soit dans les ozoneurs ou pour la destruction des polluants. Le tube central peut être un fil. Dans ce cas, la configuration est celle d’une décharge couronne. L’amorçage de la décharge est défini par la courbure des électrodes mais son extinction est due à la barrière diélectrique.
~
~
~
a b c
Diélectrique Electrode
Figure 7: Configurations typiques de décharges.
I.1.4.2. Configuration plasma/catalyseur
Dans la littérature, trois types de configurations sont étudiés entre plasma non-thermique et catalyseur : de type 1 où le plasma est en avant du catalyseur (POST-plasma, Figure 8) [9], de type 2 où le catalyseur est situé dans le gaz ionisé (IN-plasma, Figure 8) [10] et de type 3 où le plasma est toujours en avant du catalyseur mais en série avec un deuxième montage de type 2 (deux type 2 successifs) [11].
I.1. Partie bibliographie Electrode interne
~
Catalyseur Zone plasma GazType 1 : Catalyseur POST-plasma
Electrode externe
Electrode interne Catalyseur
Zone plasma
Gaz
Type 2 : Catalyseur IN-plasma
~
Electrode externeFigure 8: Catalyseur en position POST-plasma et IN-plasma (configuration DBD).
I.1.5. Réaction du méthane dans un plasma d’air
Dans un plasma d’air, le méthane peut réagir avec différentes espèces qui sont générées dans le plasma mais ceux sont généralement les espèces oxygénées les plus réactives. Le Tableau 2 présente quelques réactions de formation de réactifs oxygénés et leurs réactions avec le CH4 ainsi
que les constantes de vitesse associées [12].
Tableau 2: Réactions et constantes de vitesse pour la formation des radicaux oxygénés et leurs réactions avec CH4 (ATn(exp(-E/RT)). Constante de vitesse (cm3.s-1) Constante de vitesse (cm3.s-1) Réaction A n Réaction A n
O3→O2+O 7,16E-10 2 CH4+O2→CH3+HO2 4,04E+13 0
H+O2→O+OH 1,98E+14 0 O+CH4→OH+CH3 6,92E+10 1,56
H+O2+M→HO2+M 1,41E+18 -0,8 H+CH4→CH3+H2 1,33E+10 3
H+HO2→O+H2O 3,01E+13 0 OH+CH4→CH3+H2O 1,57E+7 1,83
O+H+M→OH+M 4,71E+18 -1 CH+CH4→H+C2H4 1,51E+12 0
O+HO2→OH+O2 3,25E+13 0 CH2+CH4→CH3+CH3 1,81E+5 0
OH+OH→O+H2O 1,50E+9 1,14
OH+OH+M→H2O2+M 2,89E+17 -0,76
O+H2O2→OH+HO2 9,63E+6 2
H2O+O→OH+OH 4,57E+9 1,3
I.1.6. Résultats expérimentaux liés à l’oxydation du méthane
Les résultats en termes de conversion et de puissance, pour l’oxydation du méthane par un système plasma non-thermique, présentés dans cette partie bibliographique ont été obtenus dans des conditions expérimentales différentes. Si le lecteur souhaite connaître les paramètres (composition gaz, débit, tension, type de catalyseur, température, …), celui-ci peut se référer au Tableau 3 présenté en page 19. Les recherches réalisées dans le but de comprendre l’interaction entre le plasma et le catalyseur d’oxydation du méthane ne sont qu’au stade préliminaire mais donnent des résultats encourageants. Nous verrons que les mécanismes réactionnels mis en jeu dans des mélanges binaires ou ternaires peuvent devenir très complexes.
I.1.6.1. Influence du plasma sur le CH4 seul
Des études ont montré que les produits de réaction d’un plasma de méthane [13-14] sont principalement des hydrocarbures de type C2. Le mécanisme réactionnel suivant est proposé [14]:
Une autre étude a été réalisée sur l’influence d’un diélectrique (quartz, gel de silice) sous forme de granulés dans le plasma [15]. Il s’avère que la courbe caractéristique (I,U) de la décharge est identique à celle avec le plasma seul. En revanche, le méthane atteint un maximum de conversion de 44 % en présence de quartz contre 34 % avec le plasma seul (à température ambiante). De plus, la sélectivité vis-à-vis du propane est plus importante avec le quartz. Il semble que la décharge soit modifiée avec les grains et que des processus à la surface des grains participent à la conversion du méthane. Les auteurs [15] proposent les réactions en chaîne suivantes pour la conversion du méthane :
Formation de radicaux CH3• et d’hydrogène atomique CH4 − →e CH3• + H• (impact électronique) Génération d’éthane CH4 + H•→ CH3• + H2 2CH3•→ C2H6 Génération d’éthylène C2H6 → 2CH3• C2H6 + H•→ C2H5• + H2 C2H6 + CH3• → C2H5• + CH4 C2H5•→ C2H4 + H• Génération d’acétylène C2H4 + H• → C2H3• + H2 C2H4 + CH3• → C2H3• + CH4 C2H3•→ C2H2 + H•
Les réactions chimiques issues d’un traitement par plasma DBD de deux mélange CH4/Ar et
CH4/N2 (rapport 1/2) ont été étudiées par spectrométrie de masse [16]. L’utilisation de gaz nobles
(Ar) ou de N2 mène principalement à la production de H2 et à des sous produits d’ordre en HC plus
élevé CnHm, n pouvant aller jusqu’à 9. Par contre, N2 semble diminuer la formation de HC longs
(CnHm) et conduit en plus à la production de composés carbonés azotés (C2N2, C3H5N, C4H7N,
C6H11N, C7H13N).
I.1.6.2. Influence du plasma sur un mélange CH4/O2
Une première étude sur l’influence du plasma sur un mélange CH4/air (C/O = 2/1) a été réalisée
avec un montage de type 3 en présence d’un catalyseur commercial Ni/α-Al2O3 [10]. Les réactions
d’oxydation partielle ou totale du méthane sont les suivantes : CH4 + 1/2O2Ù CO + 2H2 ΔH° = -36 kJ.mol-1
CH4 + 2O2Ù CO2 + 2H2O ΔH° = -802 kJ.mol-1
Lorsque la température augmente, la réaction est favorisée mais la sélectivité reste stable. L’eau et le CO sont les deux produits majoritaires de la réaction. La formation d’H2 est aussi détectée même
en présence d’oxygène non converti. Le catalyseur a une sélectivité envers H2 de 20 %. La
conversion chimique est fonction uniquement de l’énergie appliquée pour générer le plasma. Plus la quantité d’énergie par mole de réactif est importante, plus le taux de conversion augmente. En effet, l’augmentation de l’énergie déposée par impulsion dans le plasma augmente la densité d’électrons,
I.1. Partie bibliographie
augmente les impacts électroniques qui induisent le processus d’excitation et donc améliore la conversion des réactifs et ce sans affecter la sélectivité des produits de conversion. De plus, une diminution du temps de passage dans le plasma par une augmentation du débit entraîne une chute de la conversion du méthane et de l’oxygène. La présence du catalyseur Ni/α-Al2O3 dans la zone de
plasma favorise l’oxydation du CO en CO2. Une autre étude sur le même type de catalyseur [17]
(CH4 /O2=2) a montré que pour des températures inférieures à 200 °C, la conversion du méthane
était due au plasma (le catalyseur n’a pas d’influence sur le rendement et la sélectivité) et pour des températures supérieures à 200 °C le catalyseur favorisait la réaction de conversion du CO en CO2.
Une seconde étude a été réalisée sur un mélange (CH4/O2/Ar = 42/8/50) uniquement dans le plasma
[18]. Les produits de la réaction chimique dans le plasma sont majoritairement C2H4, C2H6,
méthanol, formaldéhyde, H2, CO, CO2 et H2O. Une augmentation de puissance améliore
l’oxydation du méthane et une diminution de puissance augmente la production de méthanol et d’aldéhyde.
L’influence du plasma dans un mélange CH4/O2 = 4 a été étudiée a basse pression (5-10 Torr) sur
une zéolithe ZSM-5 (Si/Al = 30) avec le montage de type 1. Différentes zéolithes ont été préparées par échange ionique afin d’étudier l’effet des métaux de transition [19]. L’ion hydrogène de la liaison H-ZSM a été remplacé par Fe, Ni, Co et Cu. D’une part, le Co est le meilleur candidat pour l’oxydation du méthane avec une conversion de 55 % et une sélectivité de 45 % pour les C2 (120
W). La différence provient de la morphologie du substrat après réaction catalytique. La surface de Co-ZSM-5 augmente par co-polymérisation de surface de radicaux C-H tandis que la surface de Ni-Co-ZSM-5 est oxydée. La conversion du méthane seul dans le plasma atteint 65 % (1000 W), puis 75 % (900 W) en présence d’O2 jusqu’à un maximum de 78 % (800 W) en présence d’O2 et du
catalyseur Co-ZSM-5. Les auteurs proposent un mécanisme réactionnel complexe pour l’oxydation du méthane par l’oxygène (Figure 9).
Figure 9: Diagramme des mécanismes réactionnels possibles dans le plasma et sur le catalyseur entre le méthane et l’oxygène [19].
I.1.6.3. Influence du plasma sur un mélange CH4/H2O
Le reformage du méthane par l’eau (CH4/H2O=1/3) à 300 °C a été réalisé dans un plasma sans
catalyseur [20]. Les composés en C2 et C3, l’H2, le CO, le CO2 et le coke sont les produits de
réaction. Trois types de coke sont observés au cours de la conversion du méthane. Le premier type de coke est marron foncé et est constitué de poudre de carbone. Le second est noir grisâtre et forme un enduit mou. Le troisième type est un filament de coke qui affecte fortement la stabilité du plasma
implique la formation de filaments quand la quantité d’eau n’est pas assez importante. Les auteurs ont aussi montré que plus le temps de séjour est long et plus la conversion des réactifs est importante.
Le mécanisme réactionnel proposé d’oxydation du méthane par l’eau est le suivant [20]: CH4 + H2O Ù CO + 3H2, ΔH° = +206 kJ.mol-1
CO + H2O Ù CO2 + H2, ΔH° = -41 kJ.mol-1 CH4 + 2H2O Ù CO2 + 4H2, ΔH° = +165 kJ.mol-1
I.1.6.4. Influence du plasma sur un mélange CH4/CO2
L’oxydation du méthane peut être générée par reformage sec (CO2). L’étude de l’influence du
plasma à 25 °C sur un mélange CH4/CO2 a été réalisée en présence d’un catalyseur ainsi que l’étude
de l’effet du rapport volumique entre CH4 et CO2 [21]. Quatre catalyseurs ont été utilisés dans un
montage de type 2: γ-Al2O3, La2O3/γ-Al2O3, Pd/γ-Al2O3 et Pd-La2O3/γ-Al2O3. D’une part, plus la
concentration en CO2 est importante et plus le méthane est converti et la sélectivité de tous les
composés C2 diminue (C2H2, C2H4 et C2H6). L’alumine seule convertit 43 % du méthane mais n’est
pas sélective en C2. Le catalyseur au La permet d’atteindre 73 % de sélectivité en C2 mais avec
seulement 25 % de conversion du méthane. L’addition de Pd sur un catalyseur à base de La n’améliore pas la conversion du méthane (25 %), ni la sélectivité (C2 produit majeur) mais joue un
rôle sur la distribution des C2. En effet, C2H4 devient le produit majoritaire. Les auteurs proposent le
schéma réactionnel suivant : CO2 + e-→ CO + O• CH4 + O•→ CH3• + OH• 2CH3• → C2H6 C2H6 + O•→ C2H5 + OH• C2H6 + e-→ C2H4 + H2 C2H4 + O• → produits C2H4 + e-→ C2H2 + H2 C2H2 + 3O• → 2CO + H2O
Ce mécanisme aboutirait aux réactions globales suivantes : 2CH4 + CO2→ C2H6 + CO + H2O
2CH4 + 2CO2→ C2H4 + 2CO + 2H2O 2CH4 + 3CO2→ C2H2 + 3CO + 3H2O CH4 + 3CO2→ 4 CO + 2H2O
D’autres recherches [13] ont porté à la fois sur l’influence du rapport CH4/CO2, de la puissance du
plasma et de la température. Le catalyseur utilisé dans le montage de type 2 est une zéolithe de type A ou de type X de tailles de pores respectives de 4,2 Å et 7,4 Å. D’une part, plus le débit augmente, plus la conversion diminue et la réaction est favorable à la formation des hydrocarbures légers (C2
augmente et C5 diminue). D’autre part, si la puissance du plasma augmente, la conversion du
méthane augmente, le rapport H2/CO diminue et la formation de composé en C5 est favorisée. La
température de paroi n’a pas d’effet significatif sur la conversion du CH4 entre 50 et 150 °C. Enfin,
I.1. Partie bibliographie
sélective envers les hydrocarbures légers (C2 et C4) et inhibe la formation de noir de carbone (coke)
et de film de carbone polymérisé.
Une étude plus précise pour connaître l’influence du plasma sur un mélange CH4/CO2 de rapport
variable a été réalisée [14] sans catalyseur avec un montage spécifique. Le méthane peut être ionisé ou non dans une branche du montage et le CO2 peut être aussi ionisé ou non dans une autre. Ce
montage permet quatre expériences différentes : CH4 et CO2 non ionisés, CH4 ionisé et CO2 non
ionisé, CH4 non ionisé et CO2 ionisé et enfin CH4 et CO2 ionisés. D’une part, plus le rapport
CH4/CO2 augmente et plus il y a formation de CO et de moins de coke. D’autre part, l’efficacité
énergétique atteint un maximum pour un ratio 50/50 avec la présence de micro-arcs mais est minimale pour le même ratio sans la présence de micro-arcs.
Les auteurs proposent le schéma réactionnel suivant pour l’activation et la conversion du CH4 en
présence de CO2 où * est soit un radical soit un site actif:
CH4 + CO2→ 2CO + 2H2, ΔH° = 247 kJ.mol-1 CH4 + * → CH4* → CH3* → CH2* → CH* → C* CO2 + * → CO2* => CO + O* CHx + O* → CO + H2 + 2* C* + O* → CO + 2* CHx* → C2H6, C2H4, C2H2, C3H6 CHx* + CO2→ CO + x/2H2 + * CH4 + O* → CO + 2H2 + *
I.1.6.5. Influence du plasma sur un mélange CH4/O2/H2O
La conversion du méthane dans un mélange CH4/O2/H2O a été étudiée dans le plasma en présence
d’un catalyseur de type Ni/α-Al2O3 [22]. D’une part, pour un catalyseur de type α-Al2O3 la
conversion du méthane est indépendante de la température et du débit de réactifs. La conversion augmente seulement avec l’énergie spécifique d’entrée (énergie appliquée au plasma). D’autre part, pour les expériences réalisées sans eau, le catalyseur montre différents résultats : à T > 300 °C, la conversion du méthane est observée en l’absence de plasma, à T < 300 °C, la combinaison du plasma et du catalyseur n’améliore pas la conversion du méthane comparée au plasma seul sur Al2O3. La présence de Ni permet l’oxydation du CO pour T > 300 °C. La conversion de l’oxygène
atteint très rapidement 100 % sur le catalyseur au Ni et ce pour de faibles valeurs d’énergie. Le Ni permet de diminuer de 25 % l’énergie requise pour la conversion en présence du plasma seul. Enfin, la combinaison de l’oxygène et de la vapeur d’eau semble jouer un rôle significatif dans l’activation du catalyseur à basse température. Cet effet peut être relié à l’adsorption de l’eau sur la surface du catalyseur en dessous de 300 °C. Cependant, la réaction étant relativement lente, il faut un temps de contact assez long pour accroître la quantité de produits oxydés. L’ajout de Ni n’améliore pas la réactivité de l’eau.
I.1.6.6. Influence du plasma sur un mélange CH4/O2/CO2/NO
L’effet d’un système plasma DBD associé à un catalyseur Al2O3 en position POST-plasma sur un
mélange complexe de type CH4/O2/CO2/NO a été étudié [23]. En absence du catalyseur, la
conversion du CH4 augmente avec l’énergie déposée. La température de light-off (T50) diminue de
470 °C pour une énergie de 36 J.L-1 à 430 °C pour une énergie de 80 J.L-1. Par contre, à ces températures, il y a formation de NOx. Afin d’éviter ces composés, il est nécessaire de diminuer à la
![Figure 5: Évolution des températures des électrons Te et des particules lourdes Tg avec la pression dans un plasma d'arc (décharge à courant continu) [5].](https://thumb-eu.123doks.com/thumbv2/123doknet/8058808.270183/26.892.263.634.605.853/figure-évolution-températures-électrons-particules-lourdes-pression-décharge.webp)
![Figure 9: Diagramme des mécanismes réactionnels possibles dans le plasma et sur le catalyseur entre le méthane et l’oxygène [19]](https://thumb-eu.123doks.com/thumbv2/123doknet/8058808.270183/30.892.218.681.652.947/figure-diagramme-mécanismes-réactionnels-possibles-catalyseur-méthane-oxygène.webp)