MARQUEURS COGNITIFS DU DÉVELOPPEMENT D'UNE MALADIE NEURODÉGÉNÉRATIVE DANS LE TROUBLE COMPORTEMENTAL EN
SOMMEIL PARADOXAL
THÈSE PRÉSENTÉE
COMME EXIGENCE PARTIELLE DU DOCTORAT EN PSYCHOLOGIE
PAR
DAPHNÉ GÉNIER MARCHAND
Avertissement
La diffusion de cette thèse se fait dans le respect des droits de son auteur, qui a signé le formulaire Autorisation de reproduire et de diffuser un travail de recherche de cycles supérieurs (SDU-522 - Rév.1 0-2015). Cette autorisation stipule que «conformément à l'article 11 du Règlement no 8 des études de cycles supérieurs, [l'auteur] concède à l'Université du Québec à Montréal une licence non exclusive d'utilisation et de publication de la totalité ou d'une partie importante de [son] travai,l de recherche pour des fins pédagogiques et non commerciales. Plus précisément, [l'auteur] autorise l'Université du Québec à Montréal à reproduire, diffuser, prêter, distribuer ou vendre des copies de [son] travail de recherche à des fins non commerciales sur quelque support que ce soit, y compris l'Internet. Cette licence et cette autorisation n'entraînent pas une renonciation de [la] part [de l'auteur] à [ses] droits moraux ni à [ses] droits de propriété intellectuelle. Sauf entente contraire, [l'auteur] conserve la liberté de diffuser et de commercialiser ou non ce travail dont [il] possède un exemplaire.»
Et voilà, soudainement, la fin de mes années universitaires est palpable! Cette thèse représente pour moi la preuve tangible d'un grand accomplissement qui n'aurait pu être possible sans l'implication de plusieurs personnes qui m'ont été très chères tout au long de ce parcours académique. Je souhaite tout d'abord remercier sincèrement mon directeur de thèse, Dr Jean-François Gagnon, pour avoir été aussi présent et disponible au cours de mes études. Jean-François, un immense merci pour ta supervision, tes conseils, tes encouragements, ton soutien financier et moral, et pour toutes les opportunités tout au long de mon doctorat. Grâce à toi, j'ai eu la chance de pouvoir expérimenter l'évaluation neuropsychologique en recherche clinique, 1 'enseignement, les présentations lors de congrès, ainsi que la rédaction de différents projets, et je considère que tout ceci m'a permis de m'épanouir comme future professionnelle. Merci également à Dr Jacques Montplaisir, mon co-directeur, pour votre confiance, les échanges toujours des plus pertinents et pour votre exemple de rigueur tout au long de mon doctorat.
Je me sens très choyée d'avoir pu effectuer mon cheminement dans un laboratoire aussi stimulant et pertinent que celui du CÉAMS de l'Hôpital du Sacré-Cœur de Montréal. Je tiens à remercier les membres de l'équipe du laboratoire qui font un travail essentiel et avec qui il est toujours agréable et enrichissant de partager. Un merci particulier à Dr Ronald Postuma pour votre expertise, les discussions, la relecture et la collaboration essentielle à mes deux articles de thèse. Merci à toute l'équipe de la clinique de sommeil pour leur contribution, et plus particulièrement merci à Jean, Sébastien, Gaétan, Dominique, Mireille et Catherine, pour votre aide et votre disponibilité. Merci également aux chercheurs du laboratoire, notamment à Dre Julie Carrier, pour avoir été un modèle de détermination dès mon entrée au baccalauréat, avoir été sensible à mes intérêts de recherche, rn 'avoir permis de me familiariser avec le monde passionnant du sommeil et pour m'avoir mise en contact avec Jean-François bien que cela signifiait que je quittais ton laboratoire! Merci également au corps professoral de 1 'UQAM, parmi lequel je tiens à remercier grandement Dre Isabelle Rouleau. Isabelle, merci d'avoir cru en moi, de m'avoir transmis ta passion de la clinique et merci pour ta présence
chaleureuse. Merci à mes superviseures d'internats, Dre Annie Malenfant, Dre Geneviève Sénéchal, et Dre Amélie Racette, pour votre générosité, votre douceur, et pour rn 'avoir donné confiance comme clinicienne. Bien entendu, je tiens à remercier les organismes subventionnaires, soit les IRSC et les FRQS, ainsi que NeuroQAM, la Faculté des Sciences humaines de 1 'UQAM, le Centre de recherche de 1 'Hôpital du Sacré-Coeur et le Ministère de 1 'Éducation, pour le soutien financier.
Plusieurs des souvenirs associés à mon doctorat resteront teintés des rencontres exceptionnelles que j'ai pu faire avec mes collègues, sans qui tout cet investissement aurait perdu énormément de valeur. Au fil des années, lors de multiples rencontres, séances d'étude, soirées et voyages, de vraies amitiés se sont créées et perdurent. Un merci chaleureux à Jessica et Shady (Vodka Tallinn since 2014!), et à Véronique, Sonia, Josie-Anne, Marjolaine, Frédérique et Jimmy, pour votre intelligence, votre authenticité et votre folie contagieuse. Merci également à mes collègues de stages et d'internats, particulièrement Audrey, Marjorie, Laurence, Delphine et Hélène sans qui les corridors universitaires et hospitaliers du Québec m'auraient paru très ternes. Enfin, merci à Pauline, mon coup de foudre d'amitié dès la première journée du doctorat. Pauline, je te remercie d'être arrivée dans ma vie avec toute ta fougue pour y rester, de m'avoir permis de me dépasser et d'avoir toujours été là pour moi ... Longue vie à Pau-né! Il va sans dire que pour réussir à terminer des études doctorales, ça prend tout un village! Merci également à mes très chères amies : Sarah, Catherine, Jessica, Émilie et Vanessa. Vous êtes toutes à votre façon une inspiration pour moi et vous insufflez à ma vie une grande dose de bonheur et de force. Je vous adore et souhaite à chaque femme d'avoir cette même sororité. Merci aux meilleurs parents du monde, Gilles et Nicole, pour votre amour inconditionnel, votre présence constante et votre support indéniable. Toute ma vie, vous avez été des modèles de persévérance et rn 'avez offert stabilité et sécurité. Je suis extrêmement reconnaissante pour 1 'investissement et la disponibilité dont vous avez fait preuve tout au long de ma vie. Je vous aime. Finalement, un merci infini à mon amoureux et meilleur ami, Éric. Tu devrais recevoir une médaille d'or pour souligner ton support dans les beaux et les moins beaux moments de ce marathon! Je t'aime et serai éternellement reconnaissante pour ta présence et ton talent unique de désamorcer toutes mes inquiétudes.
À mes parents, Gilles et Nicole, mes deux premiers profs.
REMERCIEMENTS ... ii
DÉDICACE ... iv
LISTE DES TABLEAUX ... viii
LISTE DES FIGURES ... x
LISTE DES ABRÉVIATIONS ... xi
RÉSUMÉ ... .-... xii
ABSTRACT ... xiv
INTRODUCTION ... 1
CHAPITRE I CONTEXTE THÉORIQUE ... 3
1.1. Le trouble comportemental en sommeil paradoxal (TCSP) ... 3
1.1.1. Description clinique et critères diagnostiques du TCSP ... 3
a) Épidémiologie ... 3
b) Description clinique ... 5
c) Critères diagnostiques ... 7
1.1.2. Pathophysiologie du TCSP ... 9
a) Architecture et mécanismes neurophysiologiques du sommeil ... 9
b) Lésions structurelles associées au TCSP ... 12
1.2. Les synucléinopathies et le TCSP ... 17 ·
1.2.1. Association avec les synucléinopathies (vs. tauopathies) ... 17
a) Fréquence et description du TCSP dans les synucléinopathies ... 17
b) Évolution du TC SPi ... 20
c) Observations histopathologiques ... 22
d) Description clinique des synucléinopathies ... 23
e) Stades des synucléinopathies ... 24
1.2.2. Anomalies et marqueurs de neurodégénérescence dans le TCSPi ... 27
b) Marqueurs, études longitudinales ... 30
1.3. La cognition dans le TCSP ... 31
1.3 .1. Le trouble cognitif léger (TCL) ... 31
1.3.2. La performance cognitive ... 34
a) Anomalies, études transversales ... 34
b) Marqueurs, Études longitudinales ... 36
1.4. Objectifs de recherche ... 39 CHAPITRE II ARTICLE 1 ... 42 Title page ... 43 2.1. Abstract ... 44 2.2. Introduction ... 45 2.3. Methods ... 46 2.4. Results ... 50 2.5. Discussion ... 54 2.6. References ... 61 CHAPITRE III ARTICLE 2 ... 74 Title page ... 7 5 3.1. Abstract ... 76 3 .2. Introduction ... 78
3.3. Material and methods ... ~ ... 80
3.4. Results ... ~ ... 85
3.5. Discussion ... 91
3.6. References ... 98
CHAPITRE IV DISCUSSION GÉNÉRALE ... 112
4.1 Sommaire des résultats principaux de la thèse ... 113
4.2.1 Identification des marqueurs cognitifs de la démence dans le TCSP ... 116
4.2.2 Pathophysiologie des changements cognitifs dans le TCSP ... 127
4.3 Forces et limites des résultats ... 134
4.4 Perspectives futures ... 137
CONCLUSION ... 139
ANNEXE A: Tests neuropsychologiques et normes utilisées ... 140
CHAPITRE II
Tableau 2.1 Baseline sociodemographic and clinical characteristics of ali RBD patients ... 66
Tableau 2.2 Baseline cognitive performance on neuropsychological tests of ali RBD patients ... 67 Tableau 2.3 Baseline sociodemographic and clinical characteristics of converted RBD patients ... 68 Tableau 2.4 Baseline cognitive performance on neuropsychological tests of converted RBD patients ... 69
Tableau 2.5 Psychometrie properties of the neuropsychological tests for detecting RBD patients who developed dementia first compared to match healthy subjects .... 70 Tableau 2.6 Psychometrie properties of the neuropsychological tests for detecting RBD patients who developed dementia first compared to parkinsonism first patients ... 71
CHAPITRE III
Tableau 3.1 Sociodemographic and clinical characteristics of patients at last follow-up (Y ear 0) ... 1 0 1
Tableau 3.2 Progression ofmarkers in prodromal dementia with Lewy bodies ... 102
Tableau 3.3 Psychometrie properties of cognitive tests (with area under the curve > 0.85) over time for detecting patients who developed dementia with Lewy bodies compared to healthy subjects ... ; ... 103
Tableau supplémentaire 3.1A-C. Performance on neuropsychological tests over time in association with later clinical diagnosis ... 1 06
Tableau supplémentaire 3.2A-C. Psychometrie properties of cognitive tests over time for detecting patients who developed dementia with Lewy bodies compared to
CHAPITRE II
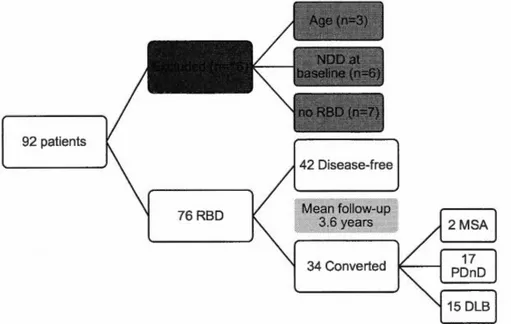
Figure 2.1 Flow ch art of study process representing inclusion criteria ... 72
Figure 2.2 Percentage of patients with impaired performance on neuropsychological tests ... 73
CHAPITRE III
Figure 3.1 Patient flowchart ... 1 04
Figure 3.2 Performance changes on neuropsychological tests (z scores) over time shawn by regression lin es ... : ... 105
En français AMS DCL EEG EOG EMG MP MPD PSG SL SP TCL TCSP TC SPi En anglais AUC DLB HC PD MCI MSA RBD REM ROC
Atrophie multi -systématisée Démence à corps de Lewy
Électroencéphalographie 1 Électroencéphalogramme Électro-oculographie 1 Électro-oculogramme
Électromyographie 1 Électromyogramme Maladie de Parkinson
Maladie de Parkinson avec démence Polysomnographie
Sommeil lent Sommeil paradoxal Trouble cognitif léger
Trouble comportemental en sommeil paradoxal
Trouble comportemental en sommeil paradoxal idiopathique
Area un der the curve
Dementia with Lewy bodies Healthy controls
Parkinson' s Disease Mild cognitive impairment Multiple system atrophy
Rapid eye movement behavior disorder Rapid eye movement
Le trouble comportemental en sommeil paradoxal (TCSP) est une parasomnie caractérisée par une perte de 1' atonie musculaire normalement associée au sommeil paradoxal. On note ainsi 1' apparition fréquente de comportements indésirables et parfois violents pouvant menacer la sécurité du patient ou du partenaire de lit. Plusieurs études ont montré un risque élevé de développer une maladie neurodégénérative chez les patients ayant reçu un diagnostic de TCSP,
particulièrement une synucléinopathie, telle que la maladie de Parkinson (MP), la démence à corps de Lewy (DCL) et l'atrophie multi-systématisée (AMS). Dans les dernières années, des anomalies structurales et fonctionnelles du cerveau, ainsi que des atteintes perceptives, motrices, autonomiques et cognitives, similaires à celles documentées dans les synucléinopathies, ont été rapportées dans le TCSP. L'objectif général de cette thèse est 1) d'identifier les marqueurs cognitifs prodromaux d'une maladie neurodégénérative et déterminer s'ils peuvent différencier les sous-types de conversion ultérieure, et 2) de suivre la progression des marqueurs cognitifs identifiés dans la période prodromale selon le type de conversion.
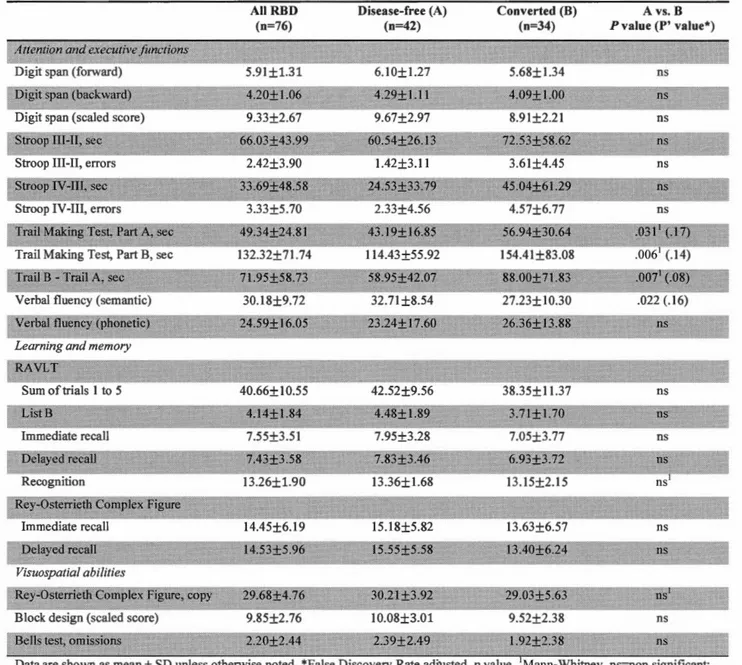
Dans la première étude, 76 patients TCSP ont été suivis sur une période moyenne de 3.6 ans. La performance initiale (TO) aux tests neuropsychologiques des patients ayant développé une synucléinopathie a été comparée à celle des patients qui étaient toujours idiopathiques au moment du suivi (Tl). Les patients ayant développé une synucléinopathie ont été séparés selon le diagnostic de parkinsonisme (MP) ou de démence à corps de Lewy (DCL). Les patients ayant développé une DCL avaient des atteintes cognitives plus sévères au TO que les patients ayant développé un
parkinsonisme ou les patients demeurés idiopathiques, ainsi qu'une fréquence plus élevée de trouble cognitif léger (TCL ). Les atteintes cognitives touchaient
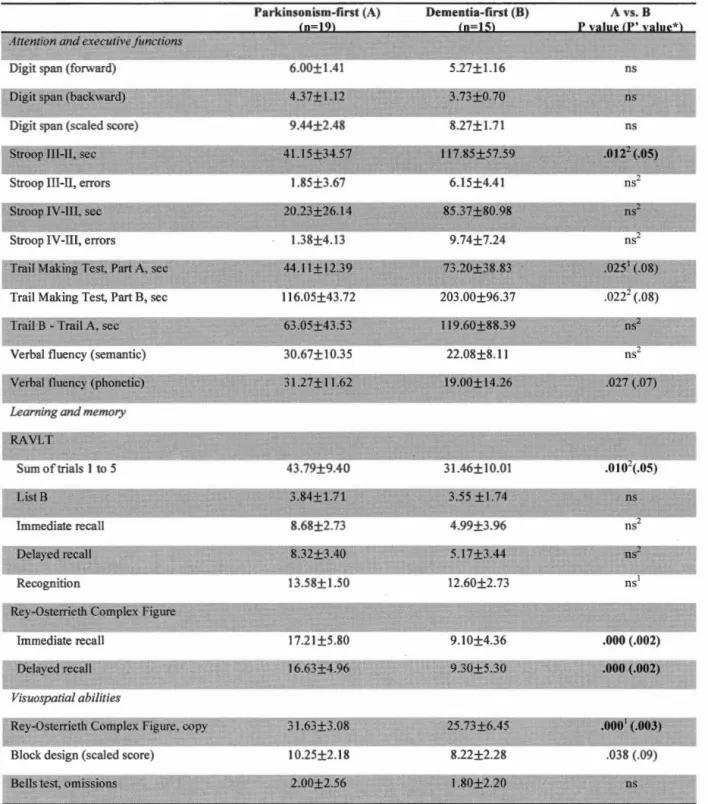
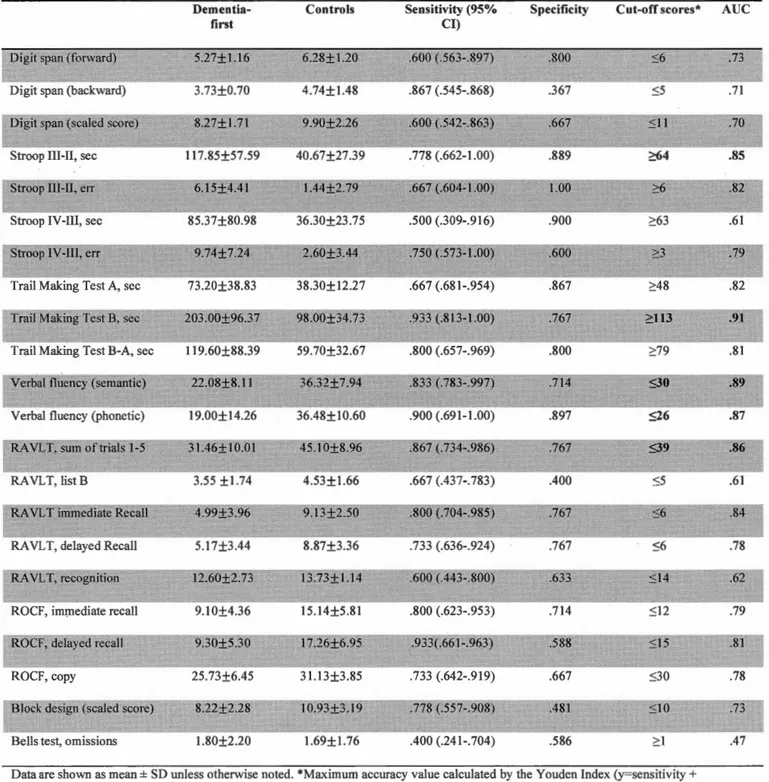
principalement l'attention et les fonctions exécutives, l'apprentissage et la mémoire épisodique et les habiletés visuospatiales. Les patients ayant reçu un diagnostic de parkinsonisme avaient une performance cognitive au TO similaire à celle des patients toujours idiopathiques. Les tâches cognitives auTO qui permettaient de prédire le développement de la DCL avec un seuil de classification supérieure à 85% étaient la tâche de Stroop (partie III, Interférence) et le Traçage de pistes (partie B, Alternance chiffres et lettres).
Dans la deuxième étude, nous avons mesuré 1' évolution des atteintes cognitives chez 109 patients TCSP dans les années précédant le diagnostic de DCL ou de MP. Trois groupes ont été définis selon le diagnostic au dernier suivi (DCL, MP ou
·idiopathique). Des analyses linéaires de modèle mixte ont été utilisées pour comparer la progression de la performance aux tests cognitifs entre les trois groupes sur une période prodromale de trois ans, et des régressions linéaires ont été effectuées sur une période prodromale de six ans pour estimer la pente de progression des trois groupes. Les changements de la performance cognitive au fil des ans étaient fortement associés au développement ultérieur de la DCL. Les atteintes attentionnelles et exécutives étaient observables au moins six ans avant le diagnostic de DCL. Les atteintes mnésiques quant à elles débutaient environ cinq ans avant, mais devenaient cliniquement significatives deux ans avant le diagnostic de DCL. Les atteintes visuospatiales ont progressé de façon variable et inconsistante. Les tâches permettant
une classification supérieure à 90% des patients qui ont développé une DCL en
comparaison à un groupe apparié de sujets sains étaient le Traçage de pistes (partie B), la fluence verbale (sémantique) et les 15 mots de Rey (total des essais 1 à 5, rappel immédiat, rappel différé).
Les résultats de cette thèse montrent qu'une atteinte précoce de 1' attention et des fonctions exécutives chez des patients avec un TCSP est associée au développement ultérieur d'une DCL. Par ailleurs, l'apparition et la progression des atteintes
mnésiques marquent la survenue imminente de la DCL.
Mots clés : Trouble comportemental en sommeil paradoxal, Neuropsychologie, Trouble cognitif léger, Maladie de Parkinson, Démence à corps de Lewy.
Rapid eye movement (REM) sleep behavior disorder (RBD) is a parasomnia characterized by Joss of normal muscle atonia during REM sleep. lt is noted that undesirable and often violent behaviors frequently emerge and could threaten safety of the patient or his bed partner. Severa} studies have shawn a high risk of developing neurodegenerative disease in patients diagnosed with RBD, especially a
synucleinopathy such as Parkinson's disease (PD), dementia with Lewy bodies (DLB) and multiple system atrophy (MSA). In the last few years, structural and functional brain anomalies as well as perceptual, motor, autonomie and cognitive anomalies similar to those observed in synucleinopathies have been reported in RBD. The purpose of this thesis is 1) to identify prodromal cognitive mark ers of a
neurodegenerative disease and to establish if they can differentiate subsequent conversion subtypes, and 2) to follow those identified cognitive markers progression in the prodromal period according to conversion subtype.
In the first study, 7 6 RBD patients were followed for a mean period of 3.6 years. The baseline cognitive performance (TO) on neuropsychological tests was compared between patients who developed a synucleinopathy and th ose who remained
idiopathie at follow-up (Tl). Patients who developed a synucleinopathy were divided in two groups according to parkinsonism (PD) or dementia (DLB) diagnoses. Patients who developed DLB had more severe cognitive impairments than patients who developed parkinsonism or patients who remained idiopathie and had a higher mild cognitive impairment (MCI) frequency. Cognitive impairments were mainly shawn in attention and executive functions, episodic learning and memory and visuospatial abilities. Patients who received a parkinsonism diagnosis had a similar cognitive performance at TOto that of idiopathie patients. At TO, the cognitive tasks that predicted the development of DLB with a classification threshold greater than 85% were Stroop Test (part III, Interference) and Trail Making Test (part B, Numbers and letters Switching).
In the second study, we systematically measured the progression of cognitive decline in 109 RBD patients during years preceding DLB or PD diagnoses. Three groups were defined accordingly to diagnosis at last follow-up (DLB, PD, or idiopathie). Linear mixed-model analyses were used to compare the progression of cognitive performance on neuropsychological tests between three groups on a three-year prodromal period, and linear regressions were executed on a six-year prodromal period to estimate the slope of progression over the prodromal period for each group. Cognitive performance changes over time were strongly associated with later
development of DLB. Deficits in attention and executive functions were observed 6 years before diagnosis. Verbal episodic learning and memory deficits started later,
deviating from normal approximately 5 years and becoming clinically impaired 2 years be fore diagnosis. Visuospatial abilities progressed variably, with in consistent prodromallatencies. The cognitive tasks that predicted the development of DLB with a classification threshold greater than 90% were the Trail Making Test (part B),
Verbal Fluency (semantic), and Rey Auditory-Verbal Leaming Test (total,
immediate, and delayed recalls).
The results of this thesis showed earl y impairments in attention and executive functions in RBD patients associated with subsequent dementia (DLB) development.
Moreover, the apparition and progression of memory impairments marks the
impending onset of DLB.
Keywords: Rapid eye movement sleep behavior disorder, Neuropsychology, Mild cognitive impairment, Parkinson's disease, Dementia with Lewy bodies
Le vieillissement pathologique représente un enJeu de société important dans le contexte socio-économique actuel. En effet, le vieillissement de la population est un phénomène graduel et inéluctable, particulièrement dans les pays occidentaux, entraînant une nécessité d'ajustements à l'organisation des soins de santé. La maladie de Parkinson (MP) et la démence à corps de Lewy (DCL) sont deux maladies neurodégénératives fréquentes qui partagent une même pathophysiologie, provoquant 1' apparition progressive de perturbations motrices, cognitives, hypniques et psychologiques. Plusieurs projets de recherche visent actuellement à identifier et à caractériser les phases précoces des maladies neurodégénératives. L'objectif est d'intervenir le plus tôt possible dans leur développement, que ce soit pour repousser 1 'apparition des symptômes, établir précocement un plan de traitement, ou améliorer la qualité de vie des patients et de leurs proches-aidants.
Le trouble comportemental en sommeil paradoxal (TCSP) est une parasomnie caractérisée par des manifestations motrices anormales durant le sommeil. Le TCSP affecterait environ 0.5% dans la population générale mais sa fréquence augmenterait à près de 5 à 7% dans la population vieillissante. Les premiers symptômes du TCSP apparaissent en moyenne entre 50 et 65 ans. Le TCSP est fortement associé aux synucléinopathies, comme la MP, la DCL, et l'atrophie 'multi-systématisée (AMS). En effet, la grande majorité des patients TCSP suivis en clinique du sommeil vont développer une synucléinopathie. Pour cette raison, ce trouble de sommeil est maintenant considéré comme un stade prodromal des synucléinopathies. De plus, plusieurs des perturbations documentées dans ces maladies neurodégénératives ont été investi guées dans le TCSP. Les études rapportent entre autres des anomalies cérébrales structurales et fonctionnelles, ainsi que des atteintes perceptives, motrices, autonomiques, cognitives et psychologiques. Le TCSP représente donc une
opportunité unique pour mieux comprendre la pathophysiologie et l'évolution de ce type de neurodégénéresence, et pour permettre d'évaluer l'efficacité des premiers traitements de neuroprotection de ces maladies neurodégénératives.
Sur le plan de la cognition, des études transversales ont montré dans le TCSP des atteintes cognitives dans les domaines des fonctions attentionnelles et exécutives, de la mémoire épisodique et des habiletés visuospatiales. De plus, une proportion élevée
de patients TCSP présentent un trouble cognitif léger (TCL ), un diagnostic
définissant un état transitionnel entre une cognition normale et la démence. À ce jour, aucune étude n'a porté sur la progression des déficits cognitifs dans le TCSP, ni sur les marqueurs neuropsychologiques pouvant permettre d'identifier les patients avec un TCSP les plus à risque de développer une synucléinopathie.
L'objectif général de la thèse est d'étudier l'évolution du profil cognitif des patients
atteints d'un TCSP idiopathique. Nous avons étudié d'abord la performance cognitive
initiale (TO) de patients ayant reçu un diagnostic de TCSP idiopathique qui ont développé ou non une synucléinopathie après un suivi moyen de 3.6 ans (Tl). Par la suite, nous avons évalué la progression des atteintes cognitives sur une période de 6 ans et identifié les valeurs psychométriques des tests cognitifs permettant de prédire le développement d'une démence (DCL). Le premier chapitre de cette thèse relate le contexte théorique ayant permis le développement des objectifs et des hypothèses de recherche. Le second chapitre présente les deux articles scientifiques de la thèse. Le premier a été publié dans la revue SLEEP en 2017 et le second dans la revue Annals of Neurology en 2018. Le troisième chapitre porte sur la discussion et la conclusion générale découlant de ces deux études. Les résultats des deux études y sont résumés, suivis de leurs implications théoriques et pratiques, ainsi que leurs limites méthodologiques. Pour conclure, des pistes de réflexion pour des projets de recherche futurs seront présentées.
CONTEXTE THÉORIQUE
1.1. Le trouble comportemental en sommeil paradoxal (TCSP)
1.1.1. Description clinique et critères diagnostiques du TCSP
a) Épidémiologie
Les parasomnies se caractérisent par l'apparition de phénomènes ou de
comportements indésirables survenant de façon prédominante ou exclusivement au
cours du sommeil (Mahowald et Schenck, 2005). Le trouble comportemental en
sommeil paradoxal (TCSP) est une parasomnie caractérisée par des manifestations motrices parfois violentes survenant au cours du sommeil paradoxal (SP). Ces comportements indésirables sont souvent liés à une activité musculaire excessive des
membres ou du menton (American Academy of Sleep Medicine, 2014; Schenck et
Mahowald, 2002). La prévalence du TCSP est estimée à moins de 0.5% dans la population générale, selon une recension téléphonique auprès de 19,961 personnes qui sondait la présence de comportements violents au cours du sommeil (Ohayon et Schenck, 201 0). À 1' aide d'une confirmation du diagnostic par enregistrement
polysomnographique (PSG), une étude populationnelle coréenne a estimé la
(Kang et al., 20 13). Des études populationnelles ont également recensé une fréquence
plus élevée oscillant entre 5 à 7% chez les personnes âgées de plus de 60-70 ans (Boot et al., 2012; Mahlknecht et al., 2015). D'autre part, le TCSP représenterait près de 5% des diagnostics effectués dans les cliniques du sommeil (Frauscher et al.,
2010).
Plusieurs études cliniques dans le TCSP ont rapporté une prévalence plus élevée chez les hommes âgés : en effet, environ 80% des patients avec un TCSP seraient de sexe masculin (lranzo et al., 2014; Oison et al., 2000; Postuma et al., 2009a; Schenck et al., 1993; Wing et al., 2008). Les causes de ce plus haut risque associé au sexe
masculin sont peu investiguées à ce jour. Selon une revue systématique de la littérature, ce ratio plus élevé chez les hommes pourrait être une surestimation due à une difficulté de détecter le TCSP chez les femmes, en raison d'une présentation distincte dans l'intensité et la nature des manifestations comportementales (Bodkin et Schenck, 2009).
La symptomatologie clinique apparait généralement entre 50 et 60 ans (St Louis et al., 20 17). Cependant, des cas à début précoce (âge < 50 ans) de TCSP sont également répertoriés; ceux-ct sont plus fréquemment associés à 1 'utilisation
d'antidépresseurs, à un historique de troubles psychiatriques, et à un ratio hommes/femmes quasi-équivalent (Ju et al., 2011; Teman et al., 2009).
-Quant à la durée de la maladie, lorsque les patients viennent consulter pour une première fois dans une clinique de sommeil, les symptômes du TCSP sont présents en moyenne depuis 7 ans (Olson et al., 2000; Postuma, et al., 2009a; Schenck et Mahowald, 2002).
b) Description clinique
Une des caractéristiques du fonctionnement normal du sommeil paradoxal (SP) se nomme l'atonie musculaire, ou la paralysie des muscles squelettiques (Jouvet, 1967). Cette fonction serait possiblement mise en place pour prévenir 1' apparition de mouvements potentiellement dangereux au cours de la survenue des rêves (Brooks et Peever, 2012; Mahowald et Schenck, 2005). L'atonie musculaire et la désynchronisation de 1' électroencéphalogramme (EEG) sont présentes tout au long du SP et en constituent les aspects toniques. Par ailleurs, les évènements phasiques surviennent de façon intermittente au cours du SP, et regroupent les saccades des mouvements oculaires rapides, les saccades musculaires du visage et des membres, une fluctuation du rythme cardiorespiratoire, ainsi que des ondes pointes
ponto-géniculo-occipitales (Montplaisir et al., 201 0).
Dans le TCSP, la perte de l'atonie musculaire observée au cours du SP peut être complète ou partielle, ce qui engendre la survenue de comportements anormaux plus ou moins complexes. Ceux-ci peuvent se manifester sous forme de vocalisations, de cris, de coups de pied ou de poing, et peuvent sembler dirigés vers un but, par
exemple pour se défendre contre un ennemi (Arnulf, 201 0; Boeve et al., 2007). Les
comportements sont parfois violents, pouvant provoquer chez le patient ou son
partenaire de lit des blessures et même mener à une consultation médicale (Schenck
et Mahowald, 2002). En effet, il est fréquent que les patients rapportent comme
complication du TCSP des blessures sur soi ou chez leur partenaire de lit de
différentes intensités, comme par exemple des chutes, des équimoses, des lacérations,
des fractures, ou même des hématomes (McCarter et al., 2014).
Lors des consultations en clinique du sommeil, les patients souffrant d'un TCSP
rapportent des rêves congruents avec les mouvements observés lors d'une nuit de
sommeil en laboratoire. En effet, la nature des comportements observables et
récurrents au cours du SP est directement associée au contenu des rêves
auto-rapportés, suggérant des manifestations comportementales mettant en action le·
scénario onirique (Oison et al., 2000; Schenck et al., 1993). D'ailleurs, plusieurs
chercheurs ont montré une association entre les comportements parfois violents
observés et le contenu de rêves auto-rapportés comme étant agressif chez les patients
avec un TCSP, incluant des thèmes de chasse, de défense ou d'attaque (Borek et al.,
2007; Fantini et al., 2005). Toutefois, il est possible que les patients aient une
tendance à se remémorer davantage un contenu de rêves et des comportements plus
violents. D'ailleurs, il se pourrait que des patients qui expérimentent des
comportements générés plus violents soient plus enclins à chercher et à recevoir de
c) Critères diagnostiques
Selon The International Classification of Sleep Disorders, Third Edition (ICSD-3),
un diagnostic de TCSP requiert de façon essentielle tous les critères suivants : A) la
présence d'épisodes récurrents de vocalisations et/ou de manifestations
comportementales complexes; B) que les comportements soient documentés soit par
une enregistrement PSG et qu'ils surviennent au cours du SP, soit par l'histoire
clinique et que cette mise en action de rêves survienne vraisemblablement au cours du
SP; C) un enregistrement PSG qui montre un SP avec des périodes sans atonie
musculaire; D) et que ce trouble ne soit pas mieux expliqué par un autre trouble de
sommeil, un trouble de santé mentale, une prise de médication, ou l'abus de
substances (American Academy of Sleep Medicine, 2014). Ainsi, bien que l'histoire
clinique permette de suspecter la présence d'un TCSP, un enregistrement PSG est
requis pour en confirmer le diagnostic.
La PSG, consistant en un enregistrement audiovisuel de différentes variables
physiologiques au cours du sommeil, est en effet essentielle pour confirmer le
diagnostic du TCSP et pour exclure d'autres troubles de sommeil pouvant mimer le
TCSP, tels que 1' apnée du sommeil, le somnambulisme, 1' épilepsie nocturne et les
mouvements périodiques des jambes (American Academy of Sleep Medicine, 2014).
La PSG permet de mesurer la présence de tonus musculaire et d'établir durant quel
stade la survenue de comportements moteurs se produit. Les anomalies observées au
musculaires toniques et phasiques lors de l'enregistrement PSG, c'est-à-dire une perte
partielle ou complète de 1' atonie musculaire et une augmentation de 1' activité
phasique des muscles du menton et des membres (Montplaisir et al., 201 0). Les
comportements anormaux surviennent habituellement plus de 90 minutes après
l'endormissement, c'est-à-dire lors de la survenue d'une période de SP, et puisque la
quantité de SP augmente au cours de la deuxième moitié de la nuit, les épisodes de
comportements anormaux dans le TCSP sont souvent plus fréquents à ce moment
(Arnulf, 201 0).
Tel que mentionné ci-haut, pour que le TCSP soit diagnostiqué via la PSG, une
augmentation de l'activité musculaire tonique et phasique doit être observée. Le seuil
exact d'une augmentation significative du tonus musculaire n'est pas
systématiquement défini dans la littérature, mais il est recommandéqu'une abolition
imparfaite du tonus musculaire soit observée pour au moins 27% du stade de SP
(American Academy of Medicine, 2014). Aussi, les autres caractéristiques du SP,
incluant la latence du SP, le pourcentage de temps en SP et le nombre de périodes de
SP, sont généralement préservées (Frauscher et al., 2012).
Les caractéristiques PSG à l'électromyogramme (EMG) pour un diagnostic de TCSP
comprennent : une activité musculaire soutenue du menton, dite tonique, et/ou une
activité musculaire transitoire, dite phasique, excessive des membres ou du menton au
cours du SP (Iber et al., 2007). Plus précisément, 1' activité tonique soutenue est
ayant une amplitude plus élevée à l'EMG que l'amplitude minimale enregistrée au cours d'une période de sommeil lent. En revanche, une activité phasique excessive consiste en des rafales musculaires d'une durée de 0.1-5.0 secondes d'une intensité au moins quatre fois plus grande que l'amplitude moyenne enregistrée à l'EMG et ce, dans au moins 50% des périodes enregistrées du SP (Iber et al., 2007).
1.1.2. Pathophysiologie du TCSP
a) Architecture et mécanismes neurophysiologiques du sommeil
Le sommeil est un état physiologique global, régi par une multitude de mécanismes et de systèmes neuronaux régulant le contrôle moteur, la vigilance, les fonctions autonomiques, le comportement et la cognition, tout comme à 1' éveil (Pace-Schott,
2002). Le sommeil se divise en deux périodes, qui se distinguent notamment par la
présence ou 1' absence de mouvements oculaires rapides, et qui se nomment
respectivement le SP (ou REM sleep, i.e. rapid eye movement sleep), et le sommeil lent (SL ou N-REM sleep, i.e. non-rapid eye movement sleep); la période du SL se
sous-divise en trois stades, N1, N2 et N3, selon les critères standardisés de
l'American Academy of Sleep Medicine (Silber et al., 2007). Différents stades de sommeil sont identifiables grâce à la PSG qui enregistre plusieurs variables physiologiques par l'EEG, l'électro-oculographie (EOG) et l'EMG. L'identification des stades de sommeil se fait sur des périodes d'enregistrement de 30 secondes, en
fonction du stade occupant majoritairement le temps dans cette période
(Rechtschaffen et Kales, 1968). L'EOG et l'EMG permettent d'identifier les périodes
de SP en mesurant respectivement les mouvements oculaires et 1' activité musculaire
du menton et des membres (Rechtschaffen et Kales, 1968). Des études ont montré
que les stades de SL et de SP alternent dans chacun des quatre à cinq cycles que
comprend chaque nuit moyenne chez un jeune adulte sain (Pace-Schott et Hobson,
2002). Aussi, une prédominance du SL est observable en début de nuit, alors que plus
la nuit avance, un cycle est de plus en plus dédié aux périodes de SP.
Plusieurs systèmes neurochimiques interagissent pour générer les états d'éveil et de
sommeil. Les états d'éveil, de sommeil et la régulation des transitions entre ces deux
états sont possibles grâce aux neurones du prosencéphale basal, du pont, du
mésencéphale, et de l'hypothalamus; par l'action de neurotransmetteurs tels que
l'acétylcholine, la noradrénaline, la dopamine, la sérotonine, l'histamine et
1' orexinelhypocrétine, ces neurones génèrent des effets diffus dans différentes régions
cibles des aires corticales et sous-corticales (Espana et Scammell, 2011). D'autre part,
les neurones GABAergiques sont très impliqués dans le contrôle et le maintien du
sommeil. Des populations de neurones GABAergiques sont notamment retrouvés en
grande quantité dans le noyau ventrolatéral préoptique et dans le noyau préoptique
médian de 1 'hypothalamus; les neurones du noyau ventrolatéral préoptique jouent un rôle essentiel dans l'instauration de l'endormissement et du SL, alors que ceux du
2016). Les noyaux cellulaires responsables du cycle d'alternance des états de veille et
de sommeil se situent principalement à la frontière du mésencéphale et du pont dans
le tronc cérébral, et comprennent les noyaux tegmentaires pédonculopontins et
latérodorsaux, du raphé dorsal et du locus coeruleus (Pace-Schott et Hobson, 2002).
La formation réticulaire, qui s'étend tout au long du tronc cérébral (du bulbe
rachidien au mésencéphale jusqu'à l'hypothalamus postérieur), est nécessaire pour
générer 1' éveil, via les systèmes noradrénergiques, histaminergiques,
sérotoninergiques et dopaminergiques, qui ont généralement des effets excitateurs et
modulateurs sur les neurones cibles afin de promouvoir l'éveil et d'amplifier des
influx nerveux excitateurs ou inhibiteurs (Espana et Scammell, 2011 ). Un autre
système neurochimique essentiel à l'éveil est l'acétylcholine. De larges groupes de
neurones cholinergiques sont retrouvés dans le prosencéphale basal et le tronc
cérébral, ainsi que dans les noyaux tegmentaires pédonculopontins et latérodorsaux
du pont (Espana et Scammell, 2011 ).
Parmi les systèmes neurochimiques actifs à l'éveil, celui de l'acétylcholine est le seul
à également présenter un taux de décharge élevé au cours du SP (Espana et
Scammell, 2011). L'activité des neurones thalamiques, qui génèrent certains rythmes
corticaux dont les fuseaux de sommeil au cours du SL via des connexions réciproques
avec le cortex, est diminuée durant le SP par 1 'action des neurones cholinergiques
mésopontins (Hu et al., 1989; Steriade et al., 1987). Ainsi, le système cholinergique
favoriser une activité corticale désynchronisée (i.e. représentée par des ondes rapides
et de faible amplitude) dans le SP similaire à celle observée au cours de l'éveil (Hu et
al., 1989).
En somme, des cellules cholinergiques du prosencéphale basal et des noyaux
tegmentaires pédonculopontins et latérodorsaux sont activées de façon préférentielle
au cours du SP, alors que d'autres cellules, comme les neurones aminergiques, ne
sont pas actives dans le SP mais actives au cours du SL et permettent alors
1 'inhibition des cellules cholinergiques. Des études plus récentes suggèrent que
l'influence mutuelle de ces deux groupes de neurones serait modulée par des circuits
inhibiteurs et excitateurs impliquant des cellules GABA et glutamatergiques (Espana
et Scammell, 20 Il; Pace-Schott et Hobson, 2002).
b) Lésions structurelles associées au TCSP
Le SP se définit et se différencie donc du SL par la présence de trois signes
cardinaux : 1) une activité EEG similaire à celle de 1' éveil, caractérisée par des
rythmes EEG de haute fréquence et de faible amplitude, 2) une atonie musculaire
presque complète du menton et des membres observée à l'EMG, et 3) des
mouvements oculaires rapides phasiques (Aserinsky et Kleitman, 1953;
Bien que l'imagerie mentale puisse être présente dans tous les stades de sommeil, le
rêve classique, caractérisé par des images vivides et un contenu émotionnel parfois
intense, se produit principalement durant le SP. L'activation des neurones réticulaires
durant le SP excite les noyaux générateurs de patrons moteurs du tronc cérébral
sous-tendant les rêves et les comportements oniriques, et ces comportements moteurs sont
normalement restreints par 1' atonie musculaire en place durant ce stade de sommeil
(Lapierre et Montplaisir, 1992).
La régulation du SP implique plusieurs systèmes dans le tronc cérébral, le
prosencéphale basal et l'hypothalamus (Fraigne et al., 2015). Plusieurs noyaux dans
la région du pont sont impliqués dans le maintien et la génération du SP, notamment
le noyau subcoeruleus. L'atonie musculaire du SP serait quant à elle contrôlée par la
formation réticulaire bulbaire (Brooks et Peever, 2012; Fraigne et al., 2015; Jouvet,
1967; Lu et al., 2006; Luppi et al., 2011; Peever et al., 2014). D'autres régions ont
montré une implication dans la régulation du SP, notamment le thalamus, la
substance noire et le cortex frontal (Jouvet, 1967; Sakai et al., 1979).
Les mécanismes et les réseaux neuronaux précis sous-jacents à la génération de
l'atonie musculaire au cours du SP sont encore aujourd'hui incertains. Chez le rat, il a
été montré que les motoneurones du nerf trijumeau innervant les muscles de la
mâchoire sont inactivés durant le SP par une transmission GABAergique et
glycinergique qui provoque l'inhibition en ciblant les récepteurs GABAA, GABAs et
cellules du noyau subcoeruleus, qui sont davantage activées au cours du SP que durant le SL. En effet, le noyau subcoeruleus regroupe des neurones glutamatergiques qui ont des projections excitatrices sur les récepteurs GABAergiques et glycinergiques des neurones du bulbe rachidien ventromédian, qui eux projettent et inhibent les neurones moteurs des muscles squelettiques. Ce circuit inhibiteur permet de produire une paralysie au cours du SP et de restreindre les saccades musculaires et les mouvements générés par les projections excitatrices du noyau rouge sur les neurones moteurs (Fraigne et al., 2015; Lu et al., 2006). En plus de l'implication du
système glutamatergique, des neurones cholinergiques du tegmentum
pédonculopontin et latérodorsal activés au cours du SP seraient également impliqués dans l'initiation et le maintien du SP et dans le contrôle de l'atonie musculaire. Ils auraient notamment un rôle médiateur et synergique dans 1 'augmentation du signal excitateur glutamatergique (Weng et al., 2014).
À ce jour, les connaissances de la pathophysiologie derrière les mécanismes sous-jacents de la perte d'atonie au cours du SP dans le TCSP proviennent principalement
des modèles animaux (St Louis et al., 20 17). Ceux-ci ont montré que des lésions du
circuit impliqué dans la génération et le maintien de 1' atonie musculaire en SP produisent un syndrome moteur de type TCSP. Plus précisément, de petites lésions bilatérales du noyau subcoeruleus et du bulbe rachidien médian peuvent engendrer des comportements moteurs au cours du SP chez les chats, les rats et les souris (Lu et al., 2006; Schenck et Mahowald, 2002; Schenkel et Siegel, 1989).
Ces découvertes sont cohérentes avec les anomalies des circuits du tronc cérébral
associées au contrôle du SP qui sont retrouvées chez les patients TCSP, notamment
dans le complexe coeruleus/subcoeruleus et les noyaux gigantocellulaire et
magnocellulaire (Ehrminger et al., 2016; Iranzo et al., 2013; Peever et al., 2014;
Scherfler et al., 2011). Dans le TCSP, une dégénérescence de ce circuit permettrait
aux projections excitatrices du cortex· moteur d'activer les motoneurones spinaux et
donc d'amplifier les évènements phasiques (i.e. saccades oculaires et contractions
musculaires) et générer des comportements moteurs complexes durant les périodes de
SP (Clement et al., 2011). Ces différentes observations confirment l'implication des
neurones glutamatergiques du noyau subcoeruleus dans la génération de 1' atonie
musculaire au cours du SP. D'ailleurs, la déprivation de neurones dans le noyau
subcoeruleus semble corréler fortement avec le SP sans atonie chez les patients ayant
une MP et un TCSP concomitant, mais pas chez les patients ayant seulement une MP
(Garcia-Lorenzo et al., 2013). D'autres structures cérébrales seraient impliquées dans
différents circuits modulant 1 'apparition de ces comportements, notamment la
substance grise périaqueducale ventrolatérale, le locus coeruleus, le noyau du raphé
dorsal, la substance noire, 1 'hypothalamus, le thalamus, 1' amygdale et le
mésencéphale (Luppi et al., 2011 ).
Le système cholinergique est également altéré dans le TCSP. Une étude de
neuroimagerie fonctionnelle a montré une désafférentation des systèmes
néocorticales, limbiques et thalamiques; cette détérioration neurochimique est associée à la présence de symptômes cliniques auto-rapportés de TCSP chez des
patients MP (Kotagal et al., 2012). Une implication du système dopaminergique dans
la perte de 1' atonie musculaire a également été raportée chez les singes, sans toutefois provoquer la génération de mouvements manifestes durant le SP (Verhave et al., 2011).
En résumé, les dysfonctions du SP observées dans le TCSP seraient dues à une dégénérescence des structures pontiques et bulbaires responsables du contrôle moteur, incluant entre autres le complexe coeruleus/subcoeruleus et la formation réticulaire (Boeve, 2013; Iranzo, 2013; Manni et al., 2013). Le TCSP existe en 1' absence de tout autre trouble neurologique, sous sa forme idiopathique (TCSPi). Des lésions structurelles causée par différentes conditions neurologiques (maladie cérébro-vasculaire, tumeurs, etc.) spécifiques aux régions du tronc cérébral peuvent également causer un TCSP symptomatique (Boeve, 20 13). En fait, une dégénérescence neuronale ou une atteinte lésionnelle dans les régions cérébrales qui régulent la suppression du tonus musculaire générant la paralysie des muscles squelettiques durant le SP peut causer un TCSP (Boeve et al., 2007). En raison des régions cérébrales impliquées, des étiologies possibles du TCSP incluent également certaines maladies neurodégénératives de la classe des synucléinopathies.
1.2. Les synucléinopathies et le TCSP
1.2.1. Association avec les synucléinopathies (vs. tauopathies)
a) Fréquence et description du TCSP dans les synucléinopathies
En raison d'une pathophysiologie commune, le TCSP est très fortement associé aux
synucléinopathies qui sont des maladies neurodégénératives caractérisées par des
dépôts intraneuronaux de la protéine alpha-synucléine (Boeve et al., 2013; Boeve et
al., 2001; Iranzo et al., 2014; McCarter et al., 2012, 2013b; American Academy of
Sleep Medicine, 2014; Molano et al., 2010; Postuma et al., 2009a; Postuma et al.,
2009b; Schenck et al., 2013). Le TCSP est aussi considéré comme un facteur de
risque majeur pour le développement des synucléinopathies, que ce soit vers un
phénotype initial de parkinsonisme ou de trouble neurocognitif majeur/démence
(Iranzo et al., 20 14; Postuma et al., 2009a; Schenck et al., 20 13).
Chez les patients ayant reçu un diagnostic de synucléinopathie, le TCSP est fréquent :
entre 30 à 4 7% des patients avec une MP (Gagnon et al., 2002; Gong et al., 20 14;
Plomhause et al., 2013; Rolinski et al., 2014; Sixel-Doring, 2011), environ 80% des
patients avec une DCL (Pao et al., 2013) et près de 90% des patients souffrant d'une
AMS (Palma et al., 20 15). En raison de son fort lien avec la DCL, le TCSP fait
maintenant partie des caractéristiques centrales permettant un diagnostic d'un trouble
neurocognitif majeur de type DCL dans la cinquième édition du Diagnostic and
dans les lignes directrices du consortium sur la DCL (Mc Keith et al., 20 17). Il a
d'ailleurs été montré que la présence du TCSP dans la DCL est associée à un début de symptômes parkinsoniens et d'hallucinations visuelles plus précoce (Dugger et al.,
2012).
Plusieurs évidences indiquent que la présence d'un TCSP chez les patients avec la MP est liée à un phénotype distinct, caractérisé par une prédominance du sexe masculin, le sous-type akinéto-rigide (i.e. rigidité/raideur et difficulté d'exécuter certains mouvements volontaires), un risque plus élevé de chutes, du freezing (i.e.
suspension involontaire de la marche), des dysfonctions autonomiques, une humeur plus déprimée, une perte olfactive et une somnolence diurne plus sévères, et la présence d'hallucinations visuelles (Gong et al., 2014; Iranzo, 2013; Neikrug et al.,
2014; Postuma et al., 2012a; Postuma et al., 2009b; Rolinski et al., 2014; Romenets et al., 20 12; Zhang et al., 20 16). On rapporte également chez les patients MP avec un
TCSP un âge plus avancé, une durée plus longue de la maladie et un pronostic plus négatif (Arnulf, 2012; Jennum et al., 2013; McCarter et al., 2013a). Enfin, la
présence d'un TCSP dans la MP est associée à des altérations cérébrales plus sévères observables à 1' imagerie fonctionnelle et anatomique (An sari et al., 2017; Arnaldi et al., 2016; Boucetta et al., 2016; Ford et al., 2013; Gagnon et al., 2004; Gaudreault et al., 2013; Kotagal et al., 2012; Limet al., 2016; Postuma et al., 2015b; Rahmani et al., 20 16; Salsone et al., 2014 ), à une performance inférieure dans les domaines
la DCL (Arnaldi et al., 2016; Chahine et al., 2016; Erro et al., 2012; Gagnon et al.,
2009; Gong et al., 2014; Jozwiak et al., 2017; Marques et al., 201 0; Rolinski et al.,
2014; Sinforiani et al., 2008; V endette et al., 2007; Wang et al., 2010; Zhang et al.,
2016) et par un plus grand déclin cognitif annuel (Chahine et al., 2016). D'ailleurs,
un diagnostic de TCSP est lié à une prévalence plus élevée de trouble cognitif léger (TCL), et prédit le risque éventuel du développement d'une démence chez les patients souffrant de la MP (Aarsland et Kurz, 2010; Anang et al., 2014; Marion et al., 2008;
Nomura et al., 2013). Une étude prospective sur une période de 4 ans auprès de
patients MP avec ou sans TCSP a montré que la prévalence de TCL était significativement plus élevée dès la première évaluation chez les patients avec un TCSP concomitant, et que les patients qui ont développé une démence avaient tous un
TCSP et un TCL au temps de base (Postuma et al., 2012a).
Dans la MP sans démence, le profil neuropsychologique est variable, c'est-à-dire. qu'il peut s'avérer normal ou comprendre certaines atteintes cognitives limitées (Boeve, 2012). Une fréquence d'environ 25% du TCL dans la MP est rapportée dans la littérature scientifique, ne tenant toutefois pas compte de la présence comorbide possible d'un TCSP (Aarsland et al., 201 0; Litvan et al., 2011 ).
Dans la DCL, on observe un profil neuropsychologique bien distinct de celui dans la maladie d'Alzheimer, qui se caractérise par des atteintes plus fréquentes et plus constantes des fonctions exécutives, de 1 'attention, de la mémoire épisodique et des fonctions visuospatiales. Or, dans la maladie d'Alzheimer, l'atteinte de la mémoire
épisodique et du langage domine dans les premiers stades de la maladie. Le profil cognitif observé dans la DCL est indissociable de celui dans la MPD, et est caractérisé par des anomalies saillantes de 1' attention, des fonctions exécutives, des habiletés visuospatiales et des anomalies plus légères du langage et de la récupération en mémoire (Miller et Boeve, 20 16).
b) Évolution du TC SPi
Dans une étude populationnelle portant sur des patients âgés de 70 à 89 ans ayant un TCSP probable, c'est-à-dire diagnostiqué via un questionnaire et confirmé par le partenaire de lit, une étude rapporte un risque de développer une maladie neurodégénérative de 34% sur une période de 4 ans (Boot et al., 20 12). Dans les
études portant sur le TCSPi réalisées en clinique du sommeil, le risque de développer une synucléinopathie a été mesuré sur une période de temps variable (Claassen et al.,
2010; Iranzo et al., 2014; Iranzo et al., 2006; Iranzo et al., 2013; Postuma et al.,
2015a; Postuma et al., 2009a; Schenck et al., 2013; Schenck et al., 1996). Dans une
première cohorte suivie sur plusieurs années, plus de 80% des hommes de 50 ans et plus initialement diagnostiqués avec un TCSPi ont développé un trouble neurodégénératif (parkinsonien ou de démence) dans un intervalle moyen de 14 ans (Schenck et al., 2013). Une seconde cohorte de 174 patients TCSPi a été suivie sur
diagnostiqués avec une maladie neurodégénérative : 29 une DCL, 22 une MP, 12 un
TCL, et 2 une AMS. Le risque estimé de développer une maladie neurodégénérative
était de 33% sur 5 ans après le diagnostic du TCSP, 76% après 10 ans, 82% après 12
ans et 91% après 14 ans. Enfin, les études provenant de notre laboratoire ont rapporté
le développement d'une synucléinopathie chez 28% des patients TCSPi après un
suivi d'environ cinq ans (Postuma et al., 2009a), et chez 66% de ceux-ci après un
suivi moyen de 7.5 ans (Postuma et al., 2015a). Il semble ainsi exister une grande
variabilité dans la progression vers une synucléinopathie au sein de cette population,
mais les données actuelles suggèrent qu'une majorité de patients TCSP sont à risque
de développer une synucléinopathie au cours d'un suivi clinique.
L'ensemble des données probantes concernant le TCSP relatées ci-haut soutiennent la
conceptualisation scientifique du TCSPi comme étant un stade prodromal de
synucléinopathies (Bum et Anderson, 201 2; Dalrymple-Alford et al., 201 1 ). Un
risque presque égal de développer initialement un parkinsonisme (MP ou AMS) ou
un trouble neurocognitif (DCL) est observé dans la plupart des cohortes cliniques de
patients TC SPi (Iranzo et al., 20 14; Postuma et al., 20 15a; Schenck et al., 2013;
c) Observations histopathologiques
Renforçant l'hypothèse de la forte association entre le TCSP et les synucléinopathies,
on retrouve plutôt rarement cette parasomnie dans la présentation clinique des
tauopathies (incluant la maladie d'Alzheimer, la paralysie supranucléaire progressive,
la démence fronto-temporale et la dégénérescence corticobasale ), soit dans seulement
5% des cas de TCSP autopsiés (Boeve, 2013). De plus, lorsque le TCSP est associé à
un diagnostic de maladie d'Alzheimer, la présence concomitante d'une pathologie
synucléine doit également être suspectée (Boeve et al., 2013).
Différentes évidences pathophysiologiques de cette association avec les
synucléinopathies sont observables dans le TCSPi, du moins lors d'une présentation
typique chez les patients âgés (St Louis et al., 20 17). Un premier laboratoire de
recherche a montré à l'aide d'examens post-mortem chez trois patients initialement
TCSPi la présence d'une pathologie à corps de Lewy (Iranzo et al., 2013). Les trois
patients avaient une perte neuronale significative ainsi qu'une accumulation
pathologique de corps et de neurites de Lewy dans la substance noire pars compacta,
et avaient tous souffert de parkinsonisme au cours de leur vie. Par ailleurs, une étude
multicentrique a effectué des analyses neuropathologiques post-mortem chez des
patients TCSP et a montré que 94% d'entre eux présentaient une pathologie de type
alpha-synucléine (160/170), et que cette fréquence augmentait à 98% lorsque seuls
les patients TCSP ayant obtenu un diagnostic confirmé par PSG étaient inclus dans
d) Description clinique des synucléinopathies
Les synucléinopathies forment un spectre étendu de présentations cliniques,
regroupant la MP (avec ou sans démence), la DCL, l'AMS, et la dysautonomie pure. Brièvement, la MP est caractérisée classiquement par un syndrome clinique moteur,
le parkinsonisme. Le parkinsonisme se définit par un ralentissement psychomoteur et une diminution de 1' amplitude et de la vitesse des mouvements (bradykinésie ),
associés à un tremblement de repos et/ou à une rigidité musculaire (Postuma et al., 20 15c ). Des manifestations non-motrices sont également présentes chez la majorité des patients et peuvent même être prédominantes dans le profil clinique, telles que des troubles de sommeil, une perte olfactive, des dysfonctions autonomiques, ou des troubles psychiatriques comme des hallucinations, 1' anxiété et la dépression (Postuma et al., 2015c). Des directives cliniques ont été récemment émises pour établir un diagnostic de TCL et de trouble neurocognitif majeur dans la MP et seront détaillées dans la section portant sur la cognition de ce chapitre (Dubois et al., 2007; Litvan et al., 2012).
La DCL est la seconde forme la plus fréquente de troubles neurocognitifs dégénératifs, pouvant représenter entre 10 à 30% des cas de démences (Mueller et al., 2017; Zaccai et al., 2005). La DCL se caractérise cliniquement par un déclin cognitif progressif provoquant des perturbations dans le fonctionnement de la vie quotidienne,
qui s'accompagne par au moins deux des critères cliniques centraux suivants : a) des fluctuations de 1 'état de vigilance et la cognition, b) des hallucinations visuelles
récurrentes qui sont typiquement bien formées et détaillées, c) un TCSP pouvant
précéder le déclin cognitif, et d) un ou plusieurs aspects cardinaux du parkinsonisme
(American Psychiatrie Association, 2013; McKeith et al., 2017). D'autres
caractéristiques soutenant le diagnostic comprennent : une sensibilité aux agents
antipsychotiques, une instabilité posturale, des chutes répétées, des épisodes
d'absence ou de syncopes, des dysfonctions autonomiques (i.e. constipation, urgence
mictionnelle diurne, hypotension orthostatique), une perte olfactive, des
hallucinations dans d'autres modalités, des épisodes de délires, ou la présence d'un
trouble psychiatrique (McKeith et al., 20 17).
Il existe un grand chevauchement entre la DCL et la MP. Le critère distinguant ces
deux conditions cliniques est le moment d'apparition des symptômes cognitifs en
relation avec les symptômes parkinsoniens. Si la démence précède ou est
diagnostiquée à l'intérieur de l'année suivant l'apparition du parkinsonisme, il s'agit
d'un diagnostic de DCL, alors que si le parkinsonisme précède la démence d'au
moins une année, le syndrome diagnostiqué sera une MPD (McKeith et al., 2017;
Miller et Boeve, 20 16).
e) Stades des synucléinopathies
Le développement des synucléinopathies se fait sur un continuum de plusieurs
maladie comme telle, incluant des atteintes motrices, cognitives, psychiatriques ou autonomiques (Jennum et al., 2013). Selon le modèle de Braak (2003), on rapporte un phénotype de progression lent et systématique chez les patients souffrant de la MP. Six stades de sévérité ont été suggérés. Les stades I et II impliquent une pathologie de corps et de neurites de Lewy dans les régions olfactives (bulbe olfactif, noyau olfactif antérieur) et dans la partie caudale du tronc cérébral (noyau moteur dorsal des nerfs vague et glossopharyngien, zone réticulaire intermédiaire, noyau gigantocellulaire
réticulaire de la formation réticulaire, noyau de raphé et complexe
coeruleus/subcoeruleus). Ensuite, les stades III et IV montrent la progression des agrégats d'alpha-synucléine dans le mésencéphale, particulièrement dans la substance noire pars compacta, pour ensuite atteindre le prosencéphale basal, le cortex transentorhinal et l'hippocampe. Puis, les stades V et VI comprennent la progression des stigmates neuropathologiques dans les aires associatives sensorielles corticales, le néocortex préfrontal et prémoteur, et parfois dans les aires sensorielles et motrices primaires. Ce pattern de progression neuropathologique suggère ainsi une apparition et une aggravation des symptômes cliniques associées aux régions anatomiques atteintes débutant par des troubles olfactifs, des dysfonctions autonomiques et du sommeil, et se poursuivant par le développement de symptômes moteurs et cognitifs (Braak et al., 2003).
Un modèle plus récent de l'évolution de la distribution topographique des corps et neurites de Lewy, The Unified Staging System for Lewy Body Disorders, a permis de
classifier 1' ensemble des patients MP autopsiés ayant reçu un examen histopathologique, et propose quatre stades de sévérité (Beach et al., 2009). D'abord,
le stade I représente une pathologie confinée au bulbe olfactif. Par la suite, le stade II se divise en deux sous-groupes : le stade lia décrivant les patients avec une pathologie prédominante dans le tronc cérébral, et le stade lib représentant une pathologie prédominante dans le système limbique. Le stade III est établi au moment où une pathologie distribuée de manière relativement équivalente entre le tronc cérébral et le système limbique est observable. Enfin, le stade IV représente une progression de la pathologie jusque dans les aires néocorticales. Ce modèle théorique permettrait d'expliquer davantage la survenue de symptômes non-moteurs très fréquente dans la phase prodromale de la MP, tels que le TCSP (Adler et Beach,
2016).
Il a été montré dans plusieurs études que la distribution de la pathologie des corps de Lewy est corrélée avec le phénotype clinique observable: des corps de Lewy répandus dans le cortex sont présents dans la DCL et la MPD et sont corrélés avec le déclin cognitif (Braak et al., 2005; Hurtig et al., 2000; Irwin et al., 20 12). De ce fait,
les symptômes cliniques sont corrélés avec la distribution régionale de la pathologie de Lewy (p.ex. pathologie dans la substance noire et perte neuronale plus sévères dans la MP et la MPD que dans la DCL) (Ho et al., 2011; Lippa et al., 2007).
La progression pathophysiologique graduelle dans les régions anatomiques associées aux synucléinopathies permet d'expliquer en partie le lien existant entre le TCSP, la
DCL, 1 'AMS et la MP. En effet, celle-ci débute généralement dans les aires du tronc cérébral aussi impliquées dans la pathophysiologie du TCSP, avant l'apparition des symptômes moteurs extrapyramidaux, comme le parkinsonisme, qui sont liés à des structures cérébrales plus rostrales (Braak et al., 2003; Halliday et al., 2011). Le TCSP peut se développer avant, en même temps ou après le début de la
synucléinopathie (Oison et al., 2000), ce qui sous-tend une implication
extrapyramidale particulière à différents patrons de neurodégénérescence dans la pathogénèse du TCSP (Manni et al., 2013). Toutefois, le TCSP précède typiquement l'apparition clinique des symptômes moteurs, cognitifs ou autonomiques associés au diagnostic de synucléinopathie par plusieurs années, voire des décennies (Claassen et al., 2010).
1.2.2. Anomalies et marqueurs de neurodégénérescence dans le TCSPi
a) Anomalies, études transversales
Des études transversales ont montré des anomalies similaires à celles observées dans les synucléinopathies et ce, dans plusieurs cohortes d'individus présentant un TCSPi.
Plusieurs altérations des structures cérébrales ont été observées dans le TCSPi par l'utilisation de la neuroimagerie : une réduction de volume dans les lobes cérébelleux, la région tegmentale du pont et le gyrus parahippocampique gauche (Han yu et al.,
2012), un amincissement cortical dans plusieurs régions frontales (gyri cingulaire antérieur et paracingulaire, gyrus rectus, gyrus frontal supérieur et orbitofrontal, et cortex moteur primaire dorsolatéral) et postérieures (gyri lingual et fusiforme), une diminution du volume de matière grise dans le lobe frontal et dans le putamen (Rahay el et al., 20 15; Rahay el et al., 20 18), ainsi qu'une contraction anormale de la surface du globus pallidus (Rahay el et al., 20 18). Des changements microstructuraux de la matière blanche ont été détectés notamment dans les régions du pont et de la substance noire (Un ger et al., 201 0), tout comme une détérioration de 1' intégrité de la matière blanche dans les aires ponto-mésencéphaliques du tronc cérébral et une augmentation de la densité de matière grise dans les régions hippocampiques (Scherfler et al., 2011 ).
Des analyses fonctionnelles ont également montré une altération de 1 'activité cérébrale associée au TCSPi, similaire à celle observée dans la MP et la DCL.
D'abord, des études ont montré une désafférentation des connexions de la substance noire avec le putamen gauche, ainsi qu'avec la zone du cuneus et du précuneus dans le lobe pariéto-occipital (Ellmore et al., 20 13), tout comme une diminution du débit sanguin cérébral régional dans le précuneus, dans 1 'uncus droit et dans les hémisphères cérébelleux (Han yu et al., 2011 ). De plus, une désafférentation dopaminergique provenant de la substance noire vers le noyau caudé est rapportée dans le TCSPi et est associée à la présence du TCSP dans la MP (Amaldi et al.,
striatale ou une hyperéchogénicité de la substance noire chez 63% des patients TCSPi; ce phénomène reflète une augmentation de la présence de dépôts de fer dans la substance noire et représente un marqueur de vulnérabilité de la voie nigrostriatale favorable à un diagnostic précoce de MP (Iranzo et al., 201 0). Un ralentissement global de l'activité électrique à l'éveil a également été rapporté dans le TCSPi (Fantini et al., 2003). Enfin, des altérations de la connectivité similaires à celles observables dans la MP au sein du réseau des ganglions de la base ont été observées à
1 'état de repos en imagerie par résonance magnétique fonctionnelle (Rolinski et al., 20 16).
De plus, on rapporte des anomalies motrices subtiles (rigidité, posture, rapidité motrice, dextérité manuelle et coordination visuo-motrice) et autonomiques (pression
sanguine, dysfonctions orthostatiques, symptômes urinaires, constipation et
dysfonctions érectiles) (Fereshtehnejad et al., 2017; Postuma et al., 2009b; Postuma
et al., 2012b), une altération olfactive et de la discrimination des couleurs (Postuma et
al., 2011 ), ainsi que des performances cognitives inférieures aux tests
neuropsychologiques dans le TC SPi (Fantini et al., 2011; Ferini-Strambi et al., 2004;
Gagnon et al., 2009; Massicotte-Marquez et al., 2008; Terzaghi et al., 2008; Youn et al., 2016).