UNIVERSITE PAUL SABATIER – TOULOUSE III
U.F.R Sciences de la Vie et de la Terre
THESE
En vue de l’obtention du
DOCTORAT DE L’UNIVERSITE DE TOULOUSE
Délivrée par l’Université Toulouse III-Paul Sabatier Spécialité : Physiopathologie Moléculaire, Cellulaire et Intégrée
Présentée par
Delphine MILHAS
RÔLE DES CASPASES ET DES SPHINGOLIPIDES
DANS LA SIGNALISATION CYTOTOXIQUE DU RECEPTEUR FAS
DANS LES LYMPHOCYTES T
Soutenue le 14 Décembre 2007 devant le jury :
M. Frédéric. RIEUX-LAUCAT Rapporteur Directeur de recherche, INSERM, Paris
M. Ali BETTAIEB Rapporteur Professeur, Université de Bourgogne-EPHE, Dijon
M. Patrick LEGEMBRE Examinateur Chargé de recherche, INSERM, Bordeaux
M. Justin TEISSIE Examinateur Directeur de recherche, CNRS, Toulouse
M. Bernard DUCOMMUN Président du Jury Professeur, Université Paul Sabatier, Toulouse
Directeurs de thèse :
M. Hervé BENOIST Professeur, Université Paul Sabatier, Toulouse III
M. Bruno SEGUI Maître de conférences, Université Paul Sabatier, Toulouse III
J’adresse mes sincères remerciements aux membres du jury pour avoir accepté de juger ce travail :
Monsieur le Docteur Frédéric Rieux Laucat et Monsieur le Professeur Ali Bettaieb, je suis très honorée de l’intérêt que vous avez porté à mon travail et je vous remercie d’avoir accepté d’en être les rapporteurs.
Monsieur le Docteur Patrick Legembre, je suis très heureuse de vous compter parmi les membres du jury.
Monsieur le Professeur Justin Teissié, je tiens à vous remercier pour notre collaboration et pour votre présence dans ce jury de thèse.
Monsieur Bernard Ducommun, je vous remercie vivement d’avoir accepté de présider ce jury, malgré une invitation tardive.
Je tiens également à remercier
Le Docteur Bruno Ségui : merci pour tout ce que tu m’as enseigné pendant ces 4 années, pour ta disponibilité, ta rigueur, ta franchise, ton humour et ta passion que j’admire.
Le Professeur Hervé Benoist : merci pour votre esprit critique, vos conseils et pour vos remarques pertinentes qui ont souvent permis de soulever des discussions scientifiques enrichissantes.
Le Professeur Thierry Levade : merci pour votre accueil chaleureux dans le monde des sphingolipides, pour votre connaissance et votre perspicacité scientifique remarquables, votre disponibilité, votre générosité et votre bonne humeur à toute épreuve.
grâce à son organisation. Merci à Nathalie, Cécile et Stéphanie. Merci à tous les étudiants qui sont passés dans le laboratoire : Sabine, Claudine, Babeth, Audrey, Nicolas Carmen et Dimitri. Merci également à Jean-Claude Lepert pour son aide précieuse en cytométrie.
Merci aussi à toute l’équipe du thème 3 pour son accueil chaleureux :
Nelly pour son écoute et ses conseils, Mogens pour les chocolats de Bruxelle et le fameux gloug, Torsten pour m’avoir permis de m’entraîner à parler en anglais. Nicole pour ta connaissance et ton aide technique mais aussi pour toutes nos conversations et nos rigolades, Patricia pour ton aide précieuse et pour avoir partagé avec moi les aléas des western, Houda, « la pompom girl », pour ta bonne humeur et nos rigolades, Dani, pour tes astuces informatiques et ta gentillesse. Merci à tout ceux qui ont partagé, pour un temps, l’aquarium avec moi: Yann, Bertrand, Nargis, Bénédicte et Lucie.
Merci à Sylvain et Toto pour la bonne ambiance que vous avez pu instaurer et nos fameuses conversations. On attend toujours le calendrier !
Dans la nouvelle équipe « sphingolipidique », merci à :
Nathalie pour ta gentillesse, tes conseils scientifiques et personnels.
Steph pour m’avoir appris à dompter les sphingolipides, pour ton aide et ta bonne humeur.
Virginie merci pour ton accueil lors de mes brefs passages à l’école des ARN Jean-Pierre pour ton American accent et ton sérieux lors des réunions.
Merci au trio de choc des 3 postdoc, Caro, Fred et Blandine pour nous faire profiter de votre expérience, à nous petits thésards.
Maintenant le sifflement de Christian est remplacé par celui de Raphaël : merci pour ta bonne humeur.
Et les petits derniers, Yayha, Guillaume, Elodie, Virginie et Julie, bon courage pour la suite.
A mes pintades adorées, Sandra, Cindy, Anne et Cat, merci pour votre amitié, tous ces moments inoubliables, vos bons petits plats et nos soirées facteur, pyjama, movida et salsa et surtout pour votre soutien dans les moments difficiles.
Merci à Marie et Jean-Phi pour votre gentillesse et vos conseils. On se retrouvera aux States.
Merci à Jean-Phi pour m’avoir initié aux plaisirs du vertige et des toboggans. Merci à tous ceux que j’ai rencontré brièvement lors des stages CIES et avec qui j’ai partagé les joies et les peines de la thèse.
Merci à toute l’équipe pédagogique de biologie cellulaire pour son accueil et les conseils de tous les enseignants avec qui j’ai travaillé, en particulier Arnaud Labrousse, Raoul Mazar, et Marjorie Fanjul.
Merci à toi ma Jojo pour ta fidèle amitié.
Merci à Alice, Delphine et Flore pour tous ces moments passés sur les bancs de la fac
A toute ma famille et un hommage particulier à mon papi et à ma mamie
A papa, maman et Romain pour votre soutien, tout votre amour et pour être là toujours, pour moi.
A toi, Emilien, pour tes encouragements permanents, ton réconfort et pour avoir supporté mon stress et mes indécisions.
RESUME
La mort des lymphocytes T est un processus essentiel pour le maintien de l’homéostasie lymphocytaire et le contrôle de la réponse immunitaire. Le récepteur Fas (CD95) et son ligand FasL (CD95-L) jouent un rôle clé dans la mort des lymphocytes T. Chez l’homme, des mutations affectant Fas, FasL ou les caspases-8 et -10 sont responsables d’ALPS (Autoimmune lymphoproliferative syndromes), soulignant l’importance de ces protéines en physiopathologie. La caspase-8 a été montrée comme essentielle dans l’apoptose induite par la stimulation de Fas. Toutefois, des travaux récents suggèrent l’existence de voies de signalisation indépendantes de la caspase-8. Le céramide, un sphingolipide bioactif, joue un rôle potentiel dans l’induction de la mort cellulaire. L’objectif de notre travail a été de clarifier le rôle des caspases et du céramide dans la mort de lymphocytes T induite par FasL. Nos résultats indiquent que la caspase-10 est capable de se substituer à la caspase-8 dans la signalisation apoptotique de Fas, en clivant Bid (Bcl-2 interacting domain) et en activant la cascade des caspases dans des cellules Jurkat. De plus, nous montrons que les caspases8 et -10 jouent un rôle dans la production de céramide et dans l’induction d’une mort nécrotique, indépendamment de leur activité catalytique.
Une inhibition de l’activité de la sphingomyéline synthase (SMS), une enzyme qui convertit le céramide en sphingomyéline, est détectée en réponse à la stimulation de Fas dans des cellules sensibles mais pas dans des cellules résistantes. L’inhibition préalable de la SMS, par approche pharmacologique ou épigénétique (siRNA), favorise l’augmentation du taux intracellulaire de céramide et la mort en réponse à FasL. L’incubation de cellules Jurkat en présence d’analogues exogènes de céramide s’accompagne d’une toxicité, suggérant un rôle du céramide dans la signalisation cytotoxique de Fas.
L’ensemble de nos résultats mettent en évidence le rôle essentiel des caspases-8 et -10 dans la mort apoptotique et nécrotique des lymphocytes T induite par FasL. De plus, l’inhibition de l’activité de la SMS favorise l’accumulation intracellulaire de céramide et pourrait participer activement à la mort des lymphocytes T induite par FasL. Interférer avec le métabolisme sphingolipidique et notamment avec l’activité de la SMS, pourrait représenter une nouvelle stratégie thérapeutique capable de sensibiliser des cellules résistantes à FasL ou aux traitements anticancéreux.
ABSTRACT
Fas (CD95) engagement by FasL (CD95L) plays a crucial function in the regulation of T cell homeostasis and in the control of immune response, essentially through cell death induction in activated-T cells. In humans, gene mutations affecting either FasL, Fas or initiator caspases-8 and -10 are responsible for ALPS (Autoimmune Lymphoproliferative Syndrome). Fas cross-linking triggers activation of both caspase-dependent and -independent pathways. In contrast to caspase-8, controversy exists as to the ability of caspase-10 to mediate apoptosis in response to FasL. FasL-induced caspase-independent signaling pathway remains largely unknown. The ceramide, a bioactive sphingolipid, plays a potential role in cell death. The aim of our study was to clarify the role of caspases and ceramide in FasL-induced T lymphocyte death.
Ours results indicate that caspase-10 can substitute to caspase-8 as an initiator caspase in Fas signaling leading to Bid (Bcl-2 interating protein) processing, caspase cascade activation, and apoptosis of human leukemia Jurkat T cells. Moreover, initiator caspases-8 and -10 can trigger cell death independently of their catalytic activities, and are absolutely required for FasL-induced ceramide production and cell death, including necrosis.
The ceramide is converted to sphingomyelin (SM) by SM synthase (SMS). In Jurkat cells, FasL treatment inhibits SMS activity in a dose- and time-dependent manner. The inhibition of ceramide conversion to SM, by pharmacological approach or by siRNA, enhances FasL-induced death of Jurkat and activated T cells. Novel ceramide analogs elevate endogenous ceramide content and promote Jurkat cell death, suggesting a role of ceramide in cytotoxic Fas signaling.
Altogether our results highlight the essential role of caspases-8 and -10 in FasL-induced apoptotic and necrotic leukemia cell death. Moreover the inhibition of SM synthesis may facilitate FasL-induced ceramide increase and lymphocytes death. Inhibiting SMS activity may represent an interesting therapeutic strategy to sensitize cells to FasL or to anti-cancer treatments.
LISTE DES ABBREVIATIONS ACAD : Activated T Cell Autonomous Death
ADN (c) : Acide DésoxyriboNucléique (complémentaire) AICD : Activation-Induced Cell Death
AIF : Apoptosis Inducing Factor
ALPS : Autoimmune LymphoProliferative Syndrome Apaf-1 : Apoptotic protease-activating factor-1 APC : AlloPhycoCyanin
ASK-1 : Apoptosis Signal-Regulating Kinase-1 aSMase : Sphingomyélinase acide
ATP : Adénosine TriPhosphate AV : Annexine-V
Bcl-2 : B-cell lymphoma 2 Bid : Bcl-2 interacting domain BSA : Bovin Serum Albumin
CAPP : Ceramide-Activated Protein Phosphatase CARD : Caspase Recruitment Domain
CCTα : CTP : phosphocholine cytidylyltransférase α CD : Cluster of Differentiation
CERT : Ceramide Transfer protein CrmA : Cytokine response modifier A DAG : DiacylGlycerol
DD : Death Domain
DED : Death Effector Domain
Diablo : Direct IAP Binding protein with LOw pI DISC : Death Inducing Signaling Complex
DNA-PK : DesoxyriboNucleic Acid-Protein Kinase DNAse I : DésoxyriboNucléase I
DOC : Acide Desoxycholique DR : Death Receptor
ELISA : Enzyme-Linked ImmunoSorbent Assay EndoG : Endonucléase G
ESM : Ecart Standard de la Moyenne
FADD : Fas-Associated protein with Death Domain FITC : Fluorescein Isothiocyanate
FLICE : FADD-like ICE
FLIP : FLICE Inhibitory Protein GCS : Glucosyl Céramide Synthase
gld : generalized lymphoproliferation disease HtrA2 : High temperature requirement protein A2 IAP : Inhibitor of Apoptosis Protein
ICAD : Inhibitor of Caspase-Activated DNase ICE : Interleukin-1β-Converting Enzyme Ig : Immunoglobuline
IL6 : Interleukine-6 IP : Iodure de Propidium JNK : cJun N-terminal Kinase KO : Knock Out
MAPK : Mitogen-Activated Protein Kinase
MTT : Bromure de 3(4,5-diméthylthiazol-2-yl)-2,5-diphényltétrazolium NFκB : Nuclear Factor-κB
NK : Natural Killer
PARP : Poly(ADP-Ribose) Polymérase PBL : Peripheral Blood Lymphocytes PBS : Phosphate Buffered Saline PC : Phosphatidylcholine
PCD : Programmed Cell Death
PDMP : 1-Phenyl-2-Decanoylamino-3-Morpholino-1-Propanol PDTC : Pyrrolidine DiThioCarbamate
PE : Phycoerythrine
PHA : Phytohémagglutinine
PI3K : PhosphatidylInositol-3-Kinase
PIDD : p53-induced protein with a Death Domain PKC : Protéine Kinase C
PLAD : PreLigand binding Assembly Domain PMSF : Phenyl Methyl Sulfonyl Fluorure PP2A/1: Protéine Phosphatase 2A/1
PPMP : 1-Phenyl-2-Palmitoylamino-3-Morpholino-1-Propanol PS : Phosphatidylsérine
RAIDD : Receptor-interacting protein (RIP)-Associated ICH-1/CED-3 homologous death protein with a Death Domain
RE : Réticulum Endoplasmique RIP : Receptor Interacting Protein RNase : Ribonucléase
ROS : Reactive Oxygen Species S1P : Sphingosine-1-Phosphate SDS : Sodium Dodecyl Sulfate
SDS-PAGE : Sodium Dodecyl Sulfate PolyAcrylamide Gel Electrophoresis siRNA : small interfering Ribonucleic Acid
SK : Sphingosine Kinase
SLE : Systemic Lupus Erythematosus
Smac : Second mitochondrial activator of caspases SMase : Sphingomyélinase
SMS : Sphingomyéline Synthase SPT : Sérine Palmitoyl Transférase SVF : Sérum de Veau Fœtal TCR : T-Cell Receptor TNF : Tumor Necrosis Factor
TNFR-1 : Tumor Necrosis Factor-Receptor-I TPCK : Tosyl Phenylalanine Chloromethyl Ketone TRAIL : TNF-Related Apoptosis Inducing Ligand
SOMMAIRE
REVUE GENERALE ... 5
I- La mort cellulaire ... 6
I-1 L’apoptose ... 6
I-1-1 Caractéristiques morphologiques et biochimiques de l’apoptose ... 6
I-1-2 La machinerie apoptotique ... 7
I-2 Les caspases... 8
I-2-1 Structure et classification ... 8
I-2-2 Mode d’activation ... 8
I-2-3 Les caspases-8 et -10 et la voie extrinsèque... 10
I-2-4 Les caspases-2 et -9 et la voie intrinsèque ... 11
I-2-5 Les caspases effectrices et leurs substrats ... 13
I-3 Régulation des caspases : protéines régulatrices et inhibiteurs ... 14
I-3-1 Les inhibiteurs d’origine cellulaire ... 14
I-3-1-1 FLIP ... 14
I-3-1-2 Les IAP ... 15
I-3-2 Les inhibiteurs d’origine virale ... 15
I-3-2-1 CrmA... 15
I-3-2-2 p35 et p49... 15
I-3-3 Les inhibiteurs chimiques... 16
I-4 Les protéines de la famille Bcl-2 ... 17
I-4-1 Structure et classification ... 17
I-4-2 Mécanismes d’action sur la mitochondrie... 18
I-5 Les facteurs solubles mitochondriaux ... 19
I-6 Les autres protéases de l’apoptose... 21
I-7 La mort caspase-indépendante... 21
I-7-1 La signalisation caspase-indépendante émanant des récepteurs de mort ... 22
I-7-2 Rôle de la mitochondrie dans la mort caspase-indépendante... 23
I-7-3 Les protéases impliquées dans la mort caspase-indépendante ... 23
I-7-4 Importance de la mort caspase-indépendante en cancérologie ... 24
II- La mort des lymphocytes T... 25
II-1 Sélection thymique ... 25
II-2 Délétion périphérique et AICD (Activation-Induced Cell Death) ... 26
II-3 Pathologies associées à un défaut de la mort des lymphocytes T ... 28
II-4 Le récepteur Fas ... 32
II-4-1 Structure de Fas... 32
II-4-2 Expression et régulation de Fas ... 33
II-4-3 Le ligand de Fas ... 34
II-4-4 Signalisation apoptotique du récepteur Fas ... 34
II-4-4-1 Initiation du signal et formation du DISC ... 34
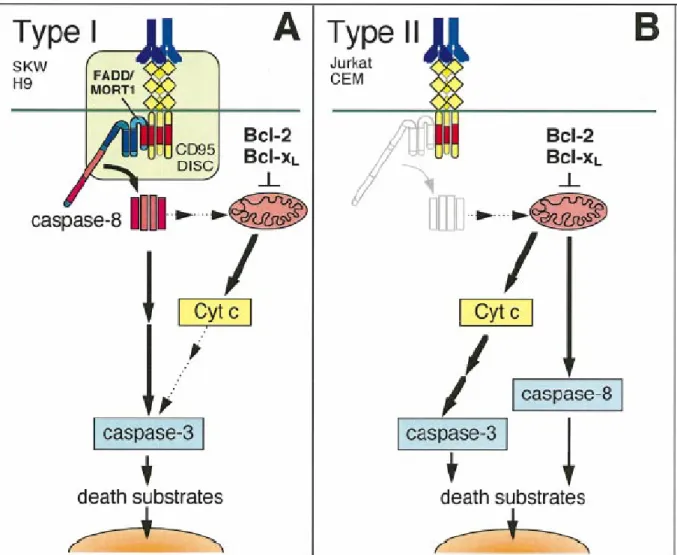
II-4-4-2 Rôle de la voie mitochondriale : cellules de type I et de type II ... 37
III-1 Structure et métabolisme des sphingolipides ... 44
III-2 Rôle des sphingolipides dans l’apoptose ... 46
III-2-1 Les métabolites pro-apoptotiques ... 47
III-2-1-1 Le céramide ... 47
III-2-1-2 La sphingosine... 49
III-2-1-3 Les gangliosides ... 50
III-2-2 Les métabolites anti-apoptotiques ... 50
III-2-2-1 Le céramide-1-phosphate ... 50
III-2-2-2 La sphingosine-1-phosphate (S1P)... 51
III-2-2-3 Le glucosylcéramide ... 51
III-2-3 Sphingolipides et morts non apoptotiques... 52
III-3 Rôle des sphingolipides dans la signalisation apoptotique du récepteur Fas... 53
III-3-1 La voie sphingomyéline-céramide dans la signalisation de Fas... 53
III-3-2 La sphingomyéline synthase (SMS) dans la signalisation de Fas ... 55
III-3-3 La synthèse de novo de céramide dans la signalisation de Fas ... 56
III-3-4 La sphingosine et la sphingosine-1P dans la signalisation de Fas... 56
III-3-5 Les glycosphingolipides dans la signalisation de Fas ... 57
III-4 Analogues sphingolipidiques et inhibiteurs du métabolisme du céramide en thérapie anticancéreuse ... 58
III-4-1 Ciblage du métabolisme du céramide... 59
III-4-2 Ciblage de la voie Sphingosine-kinase/ S1P ... 60
RESULTATS EXPERIMENTAUX ... 63
I- Introduction et objectif général ... 64
I-1 La caspase-8 n’est pas essentielle pour l’induction de l’apoptose en réponse à FasL ... 64
I-2 Existence potentielle d’une signalisation indépendante des caspases ... 65
I-3 Objectif général de la thèse ... 66
II- Rôle de la caspase-10 dans la signalisation apoptotique de Fas ... 67
II-1 Un rôle controversé de la caspase-10 dans la signalisation apoptotique... 67
II-2 Objectif et stratégie expérimentale de l’article 1 ... 67
II-3 Conclusions de l’article 1 :... 68
II-3-1 La caspase-10 est une caspase initiatrice dans la signalisation de Fas ... 68
II-3-2 Inhibition partielle de la mort cellulaire par le z-VAD... 70
II-4 Evènements mitochondriaux dans la signalisation caspase-indépendante de Fas ... 72
III- Cytotoxicité induite par les caspases-8 et -10 indépendamment de leur activité catalytique ... 75
III-1 Les cellules Jurkat déficientes en caspase-8 et -10 résistent à l’apoptose et à la nécrose induite par FasL ... 75
III-2 Objectif et stratégie expérimentale de l’article 2 ... 76
III-3 Conclusions de l’article 2... 77
III-4 L’activité catalytique des caspases est nécessaire à leur effet toxique dans les cellules HeLa... 78
IV- Rôle du céramide dans la mort de cellules Jurkat ... 80
IV-1 Production de céramide dans la signalisation de Fas... 80
IV-2 Rôle de la sphingomyéline synthase dans la signalisation de Fas ... 80
IV-3 Objectif et stratégie expérimentale de l’article 3 ... 82
IV-4-1 Inhibition de l’activité de la SMS dans la signalisation de Fas... 83
a- Inhibition dépendante de l’activité catalytique des caspases ... 83
b- Inhibition indépendante de l’activité catalytique des caspases ... 87
IV-4-2 L’inhibition de la SMS sensibilise les lymphocytes T à la mort induite par FasL... 89
IV-5 De nouveaux analogues de céramide induisent une toxicité dépendante et indépendante de l’activité catalytique des caspases ... 91
IV-5-1 Objectif et stratégie expérimentale de l’article 4... 91
IV-5-2 Conclusions de l’article 4 ... 92
DISCUSSION ... 96
I- Les caspases-8 et -10 sont indispensables à la signalisation apoptotique et nécrotique induite par FasL ... 97
II- L’inhibition de l’activité de la SMS pourrait participer à la génération de céramide dans la signalisation de Fas... 100
MATERIELS ET METHODES ... 103
I- Cultures et lignées cellulaires ... 104
I-1 Cellules Jurkat ... 104
I-2 Lymphocytes humains du sang périphérique (PBL, Peripheral Blood Lymphocytes) ... 104
I-3 Cellules HeLa ... 105
I-4 Transfections cellulaires ... 105
II- Source de FasL... 106
III- Expression de CD95 à la surface cellulaire ... 106
IV- Tests de viabilité cellulaire ... 107
IV-1 Test d’exclusion au bleu Trypan... 107
IV-2 Test MTT ... 107
V- Tests d’apoptose ... 107
V-1 Analyse morphologique ... 107
V-2 Externalisation des phosphatidylsérines et perméabilité membranaire ... 108
V-3 Analyse de la fragmentation de l’ADN... 108
V-4 Dosage de l’activité des caspases : essai enzymatique DEVDase ou IETDase... 108
V-5 Essais in vitro : protéines recombinantes ... 109
VI- Western Blot ... 109
VI-1 Extraits protéiques totaux ... 109
VI-2 Extraits protéiques cytosoliques ... 110
VI-3 Migration électrophorétique des extraits protéiques... 110
IX- Analyse de l’expression des ARNm ... 113 X- Anticorps et autres réactifs... 114
I- La mort cellulaire
Trois grands types de mort cellulaire ont été définis, l’apoptose, la mort autophagique et la nécrose. L’apoptose (PCD de type I) et la mort autophagique (PCD de type II) sont des morts cellulaires programmées (PCD, Programmed Cell Death) induites par un signal codé génétiquement. Différemment, la mort de type III, la nécrose, est provoquée accidentellement. Récemment, des formes intermédiaires de PCD caspase-indépendante ont été décrites ; il s’agit de l’apoptose-like et de la nécrose-like qui possèdent certaines caractéristiques de l’apoptose, de la nécrose et de l’autophagie. Il existe aussi deux autres types de mort cellulaire non classifiées : la catastrophe mitotique qui intervient lors d’une mitose anormale (défaut au cours de la métaphase ou de la cytodiérèse) et entraîne l’apparition respective de micro-noyaux et de cellules multinucléées et l’anoïkis qui correspond à l’apoptose induite par le détachement des cellules de leur support (Kroemer et al., 2005).
I-1 L’apoptose
L’apoptose (terme évoquant la chute des feuilles à l’automne), la plus connue des morts cellulaires programmées, a été définie en 1972. Elle correspond aux phénomènes qui conduisent à la mort physiologique des cellules. Elle a toujours été opposée à la nécrose, au cours de laquelle la rupture physique de la membrane plasmique et la destruction « brutale » de la cellule provoquées par des conditions anormales conduisent au déclenchement de réponses inflammatoires. L’apoptose joue un rôle majeur au cours du développement embryonnaire, de la croissance, ainsi que chez l'adulte, où elle est essentielle pour le maintien de l’homéostasie cellulaire. Ainsi, une dérégulation des mécanismes contrôlant l’apoptose peut participer au développement de pathologies telles que les cancers et les maladies auto-immunes (dans le cas d’un défaut de mort) ou les maladies neurodégénératives et les déficits immunitaires (dans le cas d’un excès de mort) (Fadeel et al., 1999; Thompson, 1995).
I-1-1 Caractéristiques morphologiques et biochimiques de l’apoptose
L’apoptose est définie par des modifications morphologiques caractéristiques observables au niveau du noyau et de la membrane plasmique. Une condensation et une fragmentation de la chromatine nucléaire ainsi qu’une réduction du volume cellulaire sont
d’abord observées. Puis, le noyau se fragmente et la membrane plasmique présente des évaginations (blebbing) à partir desquelles se forment plus tardivement les corps apoptotiques. Des modifications biochimiques typiques interviennent également au cours de l’apoptose : la fragmentation internucléosomale de l’ADN, un disfonctionnement mitochondrial et l’externalisation des phosphatidylsérines (PS) à la surface cellulaire. Ce dernier phénomène semble participer à la reconnaissance et à la phagocytose des cellules apoptotiques par les macrophages. Ainsi, puisque l’intégrité de la membrane plasmique est maintenue jusqu’à l’élimination des corps apoptotiques, la réaction inflammation est limitée (Kerr et al., 1972). Il est à noter qu’in vitro, des altérations de la perméabilité membranaire sont détectables aux stades tardifs ; il s’agit de nécrose secondaire ou post-apoptotique. Enfin, l’activation de protéases de la famille des Caspases (Cystéinyl aspartate specific proteinase) constitue une véritable signature de l’apoptose. En effet, les caspases clivant des protéines structurales de la cellule et inactivant des protéines essentielles à la survie, elles sont responsables de la plupart des caractéristiques morphologiques et biochimiques de l’apoptose (Cohen, 1997).
I-1-2 La machinerie apoptotique
Il existe deux voies majeures d’induction de l’apoptose :
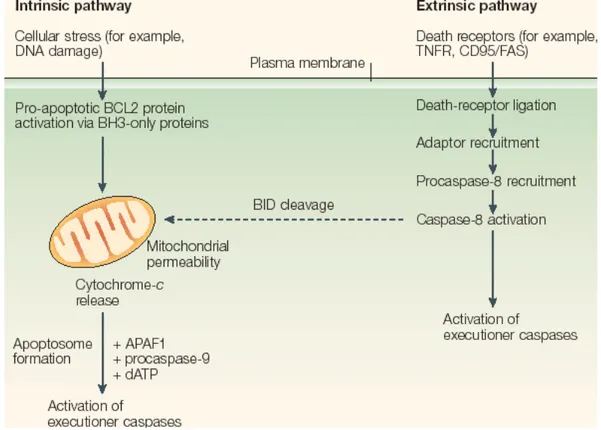
- La voie extrinsèque est initiée au niveau de la membrane plasmique par l’activation de récepteurs de mort, membres de la superfamille du TNF-R (Tumor Necrosis Factor Receptor) tels que le TNF récepteur-1 (TNFR-1), Fas (CD95 ou APO-1) et TRAIL (TNF-related apoptosis inducing ligand) récepteur, appelé aussi DR4 et DR5 pour « Death Receptor » (Smith et al., 1994). Elle permet l’activation de la cascade des caspases indépendemment de la mitochondrie.
- La voie intrinsèque implique la mitochondrie et est activée par divers agents de stress tels que la privation en sérum, les radiations ionisantes, les dommages à l’ADN, les agents chimiothérapeutiques, les infections virales ou bactériennes. Elle permet notamment l’activation de protéines membres de la famille de Bcl-2 (B-cell lymphoma 2) qui participent à la perméabilisation de la membrane externe mitochondriale (MOMP : Mitochondrial
outer-membrane mitochondriale (Figure 4) (Chipuk and Green, 2005). Au travers de la description des caspases et des protéines de la famille de Bcl-2, nous verrons comment ces deux voies sont interconnectées au cours de l’apoptose.
I-2 Les caspases
I-2-1 Structure et classification
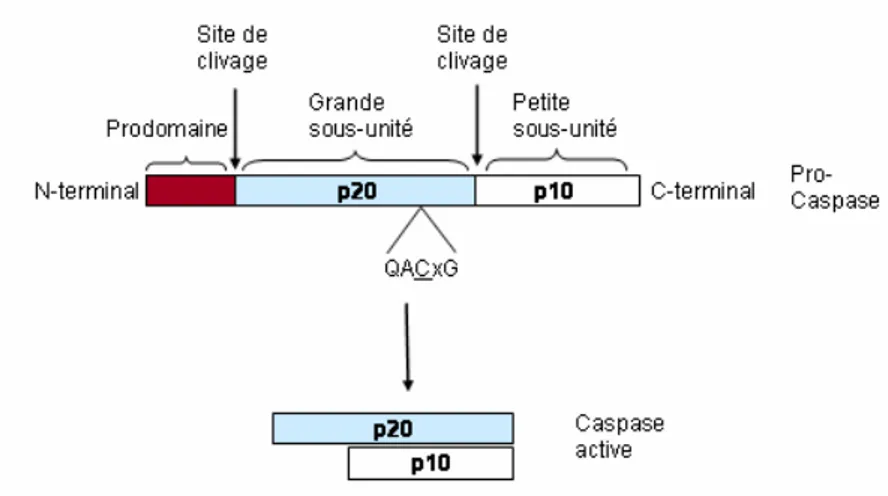
Les caspases sont des cystéines protéases possédant un résidu cystéine dans leur séquence nécessaire à l’activité catalytique. Elles clivent spécifiquement leurs substrats après un résidu d’acide aspartique. Parmi les 13 caspases identifiées chez l’homme, certaines sont impliquées dans les phénomènes d’inflammation ou de différenciation (caspases-1,-4,-5,-12 et 14) tandis que les caspases-2,-3,-6,-7,-8,-9,-10 participent à l’apoptose (Kroemer and Martin, 2005). Elles possèdent un prodomaine en N-terminal de longueur variable et un domaine catalytique en C terminal composé d’une petite sous-unité (10-12 kDa) et d’une grande sous-unité (17-20 kDa) portant le site catalytique (cf figure 1) (Fuentes-Prior and Salvesen, 2004). Les caspases de l’apoptose peuvent être divisées en deux catégories, les caspases initiatrices (caspases-2, 8, 9 et 10) possédant un long pro-domaine N-terminal et qui sont activées précocement dans la cascade apoptotique et les caspases effectrices (3, 6 et 7) qui ont un domaine N-terminal court et qui sont activées dans la phase exécutrice de l’apoptose (Thornberry and Lazebnik, 1998).
I-2-2 Mode d’activation
Elles sont synthétisées en tant que pro-enzymes inactives : sous forme de monomères pour les caspases initiatrices et de dimères pour les caspases effectrices (Boatright et al., 2003). La protéolyse au niveau de sites de clivage internes permet de libérer le site catalytique des caspases. La forme active des caspases est constituée de l’association de la petite et de la grande sous-unité formant un hétérodimère (Figure 1) (Boatright and Salvesen, 2003). Ainsi, pour les caspases effectrices le clivage protéolytique semble indispensable pour l’activation. A l’inverse, pour les caspases initiatrices la dimérisation des proformes monomériques inactives est suffisante à l’activation tandis que le clivage semble seulement stabiliser les dimères actifs formés (Boatright et al., 2003; Chang et al., 2003; Donepudi et al., 2003).
Le mode d’activation préférentiel des caspases initiatrices implique leur recrutement au niveau de plateforme de signalisation. Ce recrutement est possible grâce à des domaines particuliers présents au niveau du prodomaine des caspases initiatrices tels que le domaine CARD (Caspase Recruitment Domain) pour les caspases-9 et -2 ou le domaine DED (Death Effector Domain) pour les caspases-8 et -10 (Figure 2) (Bao and Shi, 2007). Le recrutement des caspases initiatrices au sein de ces complexes favorise leur oligomérisation et leur activation vraisemblablement par modifications conformationelles. Les caspases initiatrices une fois activées peuvent cliver et activer les caspases effectrices. De plus, il existe une boucle d’amplification positive de la cascade des caspases qui permet l’activation de caspases en amont de la signalisation par des caspases activées en aval. Par exemple la caspase-8 peut être clivée par les caspases effectrices -3 et 6 in vitro (Sohn et al., 2005).
Figure 1 : Structure générale des pro-caspases et des caspases activées. Les clivages intrachaînes permettent la séparation du prodomaine puis de la grande sous unité p20 et de la petite sous-unité p10 de la pro-caspase. La forme active des caspases est constituée de l’association de la grande et de la petite sous-unité. QACxG : séquence du site catalytique contenant le résidu cystéine.
Figure 2 : Les différents domaines structurant les caspases (Bao and Shi, 2007)
Les domaines CARD et DED sont présents au niveau du prodomaine des caspases initiatrices. Les grandes flèches représentent les sites de clivage entre les grandes (p20) et petites sous-unités (p10) tandis que les petites flèches indiquent les autres clivages. Le résidu cystéine du site catalytique est représenté par un trait rouge. Les boucles L1 à L4 forment le site actif des caspases.
I-2-3 Les caspases-8 et -10 et la voie extrinsèque
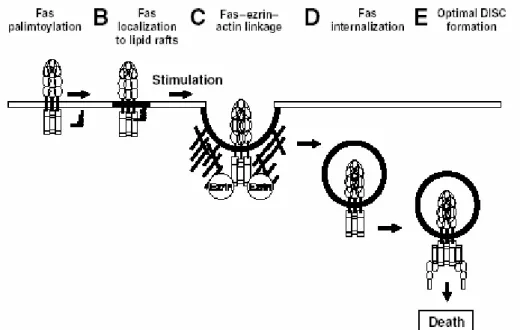
Les caspases-8 et 10 sont activées par la voie extrinsèque qui est initiée par les récepteurs de la famille du TNFR. Ces récepteurs transmembranaires de type I sont caractérisés par un domaine extracellulaire conservé riche en cystéines et un domaine intracellulaire appelé domaine de mort (DD, Death Domain) de 80 acides aminés qui permet l’interaction avec des protéines adaptatrices. Présents à la surface cellulaire sous forme de trimères pré-associés, la fixation de leurs ligands respectifs, le TNFα, FasL (CD95-L) et TRAIL (Apo2L), entraîne l’agrégation des récepteurs et leur activation permettant le recrutement de protéines adaptatrices au niveau du DD. Ainsi, dans la signalisation de Fas, la protéine FADD (Fas-Associated Death Domain protein) est recrutée au niveau du récepteur par son domaine DD (Chinnaiyan et al., 1995) et permet à son tour le recrutement des caspases-8 et -10 par le domaine DED présents à la fois sur FADD et sur les caspases initiatrices (Muzio et al., 1996). Un complexe multimoléculaire appelé DISC (Death Inducing Signaling Complex) est alors formé (Figure 3b) (Kischkel et al., 1995). Par proximité ces
caspases se dimérisent et cette dimérisation semble suffisante pour rendre les caspases actives et permettre leur maturation par protéolyse. Les clivages intrachaînes au niveau des sites consensus permettent la séparation des sous-unités catalytiques puis la libération du prodomaine. Deux hypothèses de maturation de la caspase-8 ont été émises, le clivage inter-dimères et l’autoprotéolyse (clivage intramoléculaire) (Chang et al., 2003; Chen et al., 2002). Une fois activée, les caspases-8 et -10 peuvent cliver et activer les caspases-3 et -7, des caspases effectrices de l’apoptose. Elles peuvent également induire la protéolyse d’autres protéines de la signalisation apoptotique. Par exemple le clivage de Bid (Bcl-2 interacting domain), une protéine membre de la famille de Bcl-2, par la caspase-8 permet la génération d’une protéine Bid tronquée (tBid) qui transloque à la mitochondrie et participe à la perméabilisation des membranes mitochondriales (Li et al., 1998). Ainsi, la voie extrinsèque d’induction d’apoptose rejoint la voie intrinsèque et conduit à l’induction d’événements mitochondriaux.
I-2-4 Les caspases-2 et -9 et la voie intrinsèque
Les membres de la famille de Bcl-2 participent à la voie intrinsèque en agissant sur la perméabilisation de la membrane externe mitochondriale (MOMP : Mitochondrial outer-membrane permeabilization) qui conduit au relargage du cytochrome c de la mitochondrie. Le cytochrome c libéré dans le cytosol s’associe avec Apaf-1 (Apoptotic Protease- activating factor), la protéine qui permet le recrutement ATP-dépendant de la pro-caspase-9 par l’intermédiaire des domaines CARD présents à la fois sur la caspase-9 et sur Apaf-1. Le complexe ainsi formé est appelé apoptosome et permet l’activation de la caspase-9 (Figure 3c) (Zou et al., 1999). Par ailleurs, comme son prodomaine n’est pas clivé suite à son activation, la caspase-9 reste liée à l’apoptosome au niveau duquel elle active directement la caspase-3.
La caspase-2 est également activée par la voie intrinsèque mais plus spécifiquement en réponse à des dommages à l’ADN déclenchés soit directement par les radiations ionisantes ou indirectement par l’étoposide qui, en inhibant la topoisomèrase II, favorise les cassures de l’ADN (Zhivotovsky and Orrenius, 2005). Dans ces conditions, le suppresseur de tumeur
caspase-2. Le complexe multimoléculaire formé, nommé PIDDosome, sert de plateforme d’activation pour la caspase-2 (Figure 3a) (Tinel and Tschopp, 2004). Des études in vitro indiquent qu’une fois activée la caspase-2 peut activer la caspase-8 et participer à la perméabilisation des membranes mitochondriales par l’intermédiaire du clivage de Bid (Lassus et al., 2002; Lin et al., 2004a).
Figure 3 : Les complexes d’activation des caspases initiatrices (Bao and Shi, 2007)
a, La caspase-2 est activée par le PIDDosome constitué de PIDD, RAIDD et de la
pro-caspase-2 ; b, Le DISC formé au niveau du récepteur Fas permet l’activation des caspases-8 et -10 par l’intermédiaire de FADD ; c, La caspase-9 est activée par l’apoptosome composé de sept molécules d’Apaf-1 qui se lient au cytochrome c en présence d’ATP.
Figure 4 : Les voies de signalisation intrinsèque et extrinsèque de l’apoptose conduisant à l’activation des caspases effectrices (Chipuk and Green, 2005)
I-2-5 Les caspases effectrices et leurs substrats
Activée par les caspases initiatrices, les caspases exécutrices -3, -6 et -7 achèvent la cascade apoptotique et participent à la phase terminale de l’apoptose. Ainsi elles sont responsables du phénotype apoptotique puisqu’elles clivent et dégradent de nombreux substrats essentiels à la survie cellulaire (Tableau 1). Par exemple, le clivage des lamines structurant le noyau contribue à la condensation de la chromatine. En clivant des composants ou des protéines régulatrices du cytosquelette telles que l’actine ou la gelsoline, les caspases effectrices participent à la désorganisation des structures cellulaires (Kothakota et al., 1997). En particulier, le clivage des fodrines pourrait jouer un rôle dans les phénomènes de « blebbing » membranaire. De plus, les caspases effectrices inactivent des protéines nécessaires au maintien de l’intégrité du génome, provoquant les phénomènes de fragmentation de l’ADN. Par exemple le clivage de ICAD (Inhibitor of CAD/DFF45) permet la libération de CAD (Caspase-Activated deoxyribonuclease), une nucléase responsable de la fragmentation de l’ADN. De même, le clivage de PARP (Poly-(ADP-Ribose) Polymérase) ou de la DNA-PKcs inhibe leur activité de réparation de l’ADN, favorisant les cassures de l’ADN (Cohen, 1997; Thornberry and Lazebnik, 1998).
Finalement, malgré un rôle prépondérant des caspases effectrices dans la phase d’exécution de l’apoptose, il semblerait que les caspases-3 et -7 exercent une boucle de rétrocontrôle positif permettant l’amplification des évènements mitochondriaux. En effet, des cellules déficientes à la fois en caspase-3 et en caspases-7 présentent un défaut dans l’induction des évènements mitochondriaux en réponse aux radiations UV (Lakhani et al., 2006).
Tableau 1 : Les substrats des caspases et leur séquence consensus de clivage (Cohen, 1997)
I-3 Régulation des caspases : protéines régulatrices et inhibiteurs
I-3-1 Les inhibiteurs d’origine cellulaire
I-3-1-1 FLIP
La protéine c-FLIP (cellular-FLICE inhibitory protein), l’homologue de la protéine virale v-FLIP est connue pour inhiber la signalisation apoptotique médiée par les récepteurs de mort. Elle existe sous forme de deux variants d’épissage alternatif, FLIPs (forme courte) et FLIPL (forme longue). FLIPs est constituée de deux domaines DED tandis que FLIPL est un homologue de la caspase-8 protéolytiquement inactif. Par compétition les deux formes de c-FLIP préviennent le recrutement des caspases initiatrices-8 et -10 par la protéine FADD et inhibent la formation du DISC (Irmler et al., 1997). Cependant, alors que FLIPs semble toujours agir en tant qu’inhibiteur, de faibles concentrations de FLIPL permettent l’activation protéolytique de la caspase-8 au niveau du DISC grâce à la formation de l’hétérodimère caspase-8/FLIP (Boatright et al., 2004; Chang et al., 2002). Cette observation renforce l’hypothèse selon laquelle la dimérisation des caspases initiatrices est suffisante pour favoriser leur activation.
I-3-1-2 Les IAP
Les IAPs (Inhibitor of apoptosis protein) appartiennent à une famille de 8 protéines inhibitrices de l’apoptose (Salvesen and Duckett, 2002). XIAP, la mieux caractérisée, se lie aux caspases -3, -7 et -9 grâce à un domaine conservé, BIR (Baculovirus IAP Repeat), et les inactivent (Takahashi et al., 1998). Les IAPs possèdent également un domaine RING impliqué dans la dégradation par le protéasome des IAPs elles mêmes (Yang et al., 2000) et des caspases avec lesquelles elles interagissent (Suzuki et al., 2001).
I-3-2 Les inhibiteurs d’origine virale
I-3-2-1 CrmA
CrmA (Cytokine response modifier A), une serpine (inhibiteur de sérine protéase) produite par le virus cowpox prévient les réponses inflammatoires et apoptotiques en inhibant préférentiellement l’activité des caspases-1 et -8 (Zhou et al., 1997). La fixation de CrmA sur le site catalytique des caspases entraîne l’inhibition irréversible de leur activité par dissociation du tétramère composant la caspase active et perte de la petite sous-unité catalytique (Dobo et al., 2006). Ainsi, CrmA inhibe l’apoptose induite par Fas dans des lymphocytes, suggérant un rôle important de la caspase-8 dans cette signalisation (Smith et al., 1996). Malgré son effet inhibiteur sur la caspase-9 in vitro, CrmA n’a aucun effet sur la mort médiée par la voie intrinsèque qui implique la caspase-9 (Ryan et al., 2002).
I-3-2-2 p35 et p49
La protéine p35 produite par le Baculovirus et son homologue p49 inhibent l’activité d’un spectre large de caspases incluant la caspase-1, -3, -6, -7, -8, -9 et -10 (Callus and Vaux, 2007). Le clivage de p35 par les caspases entraîne la formation d’un complexe caspase/p35 qui bloque le site catalytique des caspases (Zhou et al., 1998).
Tableau 2 : Spécificités des inhibiteurs cellulaires et viraux des caspases (Callus and Vaux, 2007)
I-3-3 Les inhibiteurs chimiques
De nombreux inhibiteurs chimiques des caspases ont été synthétisés dans le but d’étudier le rôle des caspases dans les processus de mort cellulaire ou d’inflammation. Ce sont des peptides qualifiés de pseudo substrats puisqu’ils contiennent la séquence de clivage de la caspase cible. Ils sont couplés à des groupements chimiques, le plus souvent aldéhydes (CHO) ou chloro/fluoro-méthylcétones (fmk) qui permettent non seulement d’inhiber le site actif des caspases mais aussi d’augmenter leur stabilité et leur efficacité (Callus and Vaux, 2007). Ainsi, ils agissent en tant qu’inhibiteurs compétitifs en se fixant de façon réversible (couplage CHO) ou irréversible (couplage fmk) sur le site catalytique des caspases. Certains peptides possèdent des spécificités pour des caspases particulières (Tableaux 3 et 4) (Garcia-Calvo et al., 1998) ; tandis que d’autres sont des inhibiteurs à large spectre tel que le z-VAD-fmk qui inhibe irréversiblement toutes les caspases, avec une efficacité moindre vis-à-vis de la caspase-2 (Tableau 3) (Callus and Vaux, 2007). Cependant, certains de ces inhibiteurs ne sont pas spécifiques des caspases, comme le z-VAD.fmk, le z-DEVD.fmk et le YVAD.cmk qui inhibent également l’activité, in vitro et en culture, d’ autres types de cystéine protéases comme la Cathepsine B (Schotte et al., 1999). Ainsi, l’utilisation de ces inhibiteurs pour étudier spécifiquement le rôle des caspases doit être complété par d’autres approches. Finalement, des molécules utilisables en clinique dans le traitement des maladies neurodégénératives ont été développées par différents groupes pharmaceutiques et correspondent à des inhibiteurs de caspases non peptidiques (cf tableau 4) (Callus and Vaux, 2007).
Tableau 3 : Spécificités des inhibiteurs chimiques des caspases (Callus and Vaux, 2007)
Tableau 4 : Les constantes d’inhibition (Ki) des différents inhibiteurs peptidiques des caspases (Garcia-Calvo et al., 1998).
I-4 Les protéines de la famille Bcl-2
I-4-1 Structure et classification
Les protéines de la famille de Bcl-2 représentent une vingtaine de membres, pouvant être divisés en trois sous-catégories en fonction de leur rôle dans l’apoptose et la présence de
famille Bax incluant Bax, Bak, et Bok qui possèdent trois domaines BH et les protéines à domaine unique BH3 (BH3 only) dont les plus connues sont Bid et Bad. Le domaine BH3 est nécessaire pour l’effet pro-apoptotique tandis que le domaine C-terminal hydrophobe (TM) permet un ancrage dans les membranes des organelles intracellulaires et en particulier dans la membrane externe mitochondriale. Bcl-2, Bcl-xL et Bak sont des protéines membranaires majoritairement localisées dans la membrane externe mitochondriale tandis que la plupart des autres membres de la famille Bcl-2 résident dans le cytosol et transloquent à la mitochondrie sous l’effet d’un stimulus apoptotique (Cory and Adams, 2002). Une littérature récente indique un rôle potentiel des membres de la famille de Bcl-2 dans le contrôle de la perméabilité membranaire d’autres organites comme le lysosome, le RE et le noyau.
Figure 5 : Structure des membres de la famille de Bcl-2 (Cory and Adams, 2002). Les
domaines BH1 à BH4 sont des régions conservées. Les hélices α connues sont représentées. TM, Transmembrane Domain.
I-4-2 Mécanismes d’action sur la mitochondrie
Les protéines « BH3-only » sont les sentinelles qui déclenchent la signalisation intrinsèque de l’apoptose en réponse aux stimuli de stress et qui participent aussi à la signalisation extrinsèque médiée par les récepteurs de mort. Dans des conditions normales, leur activité pro-apoptotique est réprimée. Par exemple, Bad est phosphorylée par akt/PKB et est séquestré par la protéine 14-3-3 (Zha et al., 1996). L’inhibition d’akt/PKB favorise la déphosphorylation de Bad et sa relocalisation mitochondriale. Bid est cytosolique et
transloque à la mitochondrie qu’après clivage en tBid par la caspase-8 (Li et al., 1998). Ainsi, tBid induit l’activation de Bax et de Bak qui s’oligomérisent et forment des pores à travers la membrane mitochondriale (Korsmeyer et al., 2000). Les mécanismes de formation de ces canaux diffèrent selon le type de protéines pro-apoptotiques. Ainsi, Bax et Bak peuvent être activés directement par Bid ou Bim tandis que Bad et Bik participent à la perméabilisation des membranes mitochondriales de façon indirecte en inhibant les protéines anti-apoptotiques Bcl-xL et Bcl-2 (Kuwana et al., 2005). Bcl-xL et Bcl-2 préviennent l’ouverture de ces canaux par des mécanismes qui restent encore incertains : soit par séquestration des protéines BH3-only (Cheng et al., 2001), soit par inhibition de l’activation de Bak et de Bax (Adams, 2003).
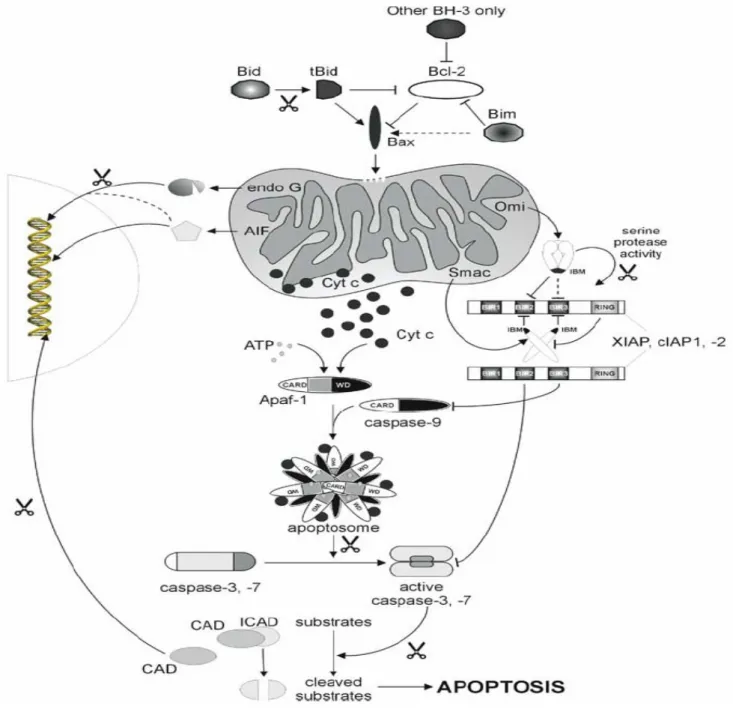
I-5 Les facteurs solubles mitochondriaux
La perméabilisation de la membrane externe mitochondriale est un évènement majeur dans la signalisation apoptotique puisqu’elle permet la libération de nombreux facteurs mitochondriaux nécessaires au processus apoptotique (Saelens et al., 2004; van Gurp et al., 2003). Tout d’abord le cytochrome c résidant dans l’espace intermembranaire mitochondrial, une fois libéré dans le cytosol participe à la formation de l’apoptosome et à l’activation de la caspase-9 (cf I-2-4) (Liu et al., 1996). De même, Smac/Diablo (Second mitochondria-derived activator of caspases / Direct IAP binding protein with low pI) et Omi/HtrA2 (High temperature requirement A2) sont également libérés de la mitochondrie et participent à l’apoptose en antagonisant les IAPs (cf I-3-1). La libération d’AIF (Apoptosis Inducing Factor) et d’EndoG (Endonucléases G) dans le cytosol et leur translocation dans le noyau conduit à la dégradation de l’ADN. En effet, AIF induit la macrofragmentation de l’ADN (génération de fragments de hauts poids moléculaires) et participe à la condensation chromatinienne, tandis qu’EndoG, en collaboration avec des exonucléases et la DNAse I, permet la dégradation internucléosomale de l’ADN. Il a également été suggéré que certaines caspases résidaient dans l’espace intermitochondrial (Zhivotovsky et al., 1999) mais cette localisation mitochondriale reste controversée (van Loo et al., 2002a).
Deux modèles pouvant coexister ont été proposés pour expliquer le mécanisme de libération des facteurs mitochondriaux : (i) la formation de canaux par les membres de la
mitochondriale interne. Les stimuli apoptotiques déclenchent l’ouverture de ces pores, induisant la dissipation du potentiel transmembranaire mitochondrial et la rupture de l’homéostasie ionique et osmotique de la mitochondrie. Ceci a pour conséquence la rupture de la membrane mitochondriale externe. De plus, plusieurs études indiquent des interactions possible entre les membres de la famille de Bcl-2 et les composants du PTP (Armstrong, 2006).
Figure 6 : Les protéines de la famille de Bcl-2 et les évènements mitochondriaux dans la signalisation intrinsèque de l’apoptose (Saelens et al., 2004).
I-6 Les autres protéases de l’apoptose
De nombreux travaux indiquent que d’autres protéases participent à la signalisation apoptotique, parmi lesquelles citons, les granzymes, les calpaïnes et les cathepsines.
Les granzymes sont des sérines protéases libérées par exocytose par les lymphocytes T et les cellules NK (Natural Killers) pour tuer les cellules cibles (cellules infectées, cellules tumorales). La granzyme B clive ses substrats après un résidu d’acide aspartique ; elle est donc capable d’induire l’apoptose notamment en clivant et en activant de nombreuses caspases telles que les caspases -3, -6, -7, -8, -9 et -10 (Johnson, 2000). Les calpaïnes (calcium activated neutral protease) sont des cystéines protéases activées par autolyse par divers stimuli apoptotiques tels que les irradiations, les agents antitumoraux, les ionophores et en particulier ceux induisant un stress du Réticulum Endoplasmique (RE), qui provoquent une augmentation du taux de calcium dans le cytosol. Elles partagent des substrats communs avec les caspases (Wang, 2000). Par exemple, les calpaïnes clivent Bax lors de l’apoptose de cellules leucémiques HL-60 induite par des drogues antitumorales (Wood et al., 1998). Les cathepsines sont des protéases lysosomales synthétisées sous forme de zymogènes et activées par protéolyse. Elles sont libérées dans le cytoplasme après perméabilisation des membranes lysosomales induite par les stimuli apoptotiques. Elles semblent activer la voie apoptotique mitochondriale ; la cathepsine D participe à l’apoptose de lymphocytes T induite par la staurosporine en activant Bax (Bidere et al., 2003).
I-7 La mort caspase-indépendante
La mort dépendante est la plus étudiée, mais des voies alternes caspase-indépendante conduisent au déclenchement de mort cellulaire en réponse à divers stimuli pro-apoptotiques. Des travaux in vitro et in vivo, en partie basés sur le fait que le z-VAD, un inhibiteur à spectre large des caspases, ne bloque pas la mort cellulaire, attestent de l’existence d’une mort indépendante des caspases : (i) lors du développement embryonnaire, la perte des cellules interdigitales peut être provoquée par un mécanisme indépendant des caspases (Chautan et al., 1999) ; (ii) l’inhibition de l’activité des caspases par le z-VAD-fmk
Une classification des différents types de morts cellulaires programmées a été proposée ; basée sur des critères morphologiques et biochimiques permettant de distinguer l’apoptose des deux types de mort caspase-indépendante, nommées « apoptose-like » et « nécrose-like ». Ainsi, la mort de type « apoptose-like » possède certaines caractéristiques de l’apoptose telles que la condensation chromatinienne et l’externalisation des PS, mais les phénomènes de blebbing, de fragmentation nucléaire, de microfragmentation de l’ADN et la formation de corps apoptotiques ne sont pas observés. La mort de type « nécrose-like » se caractérise plutôt par une augmentation de la perméabilité membranaire, un gonflement des organites intracellulaires et par l’absence de condensation chromatinienne (Jaattela and Tschopp, 2003; Kroemer and Martin, 2005; Leist and Jaattela, 2001).
I-7-1 La signalisation caspase-indépendante émanant des récepteurs de mort
Les mécanismes moléculaires impliqués dans la signalisation cytotoxique caspase-indépendante ne sont pas clairement élucidés. Des travaux du groupe de Vandenabeele indiquent que l’activation des récepteurs de mort Fas et TNFR-1 conduit à une mort caspase-indépendante impliquant la production d’espèces réactives de l’oxygène (ROS) (Vercammen et al., 1998a; Vercammen et al., 1998b). La sérine thréonine kinase RIP1 (Receptor Interacting Protein) semble nécessaire à la mort caspase-indépendante médiée par le TNFR-1, Fas et TRAIL, puisque la déficience en RIP, ou une diminution de son expression, inhibe la mort induite par l’activation de ces trois récepteurs en présence de z-VAD (Holler et al., 2000; Thon et al., 2006). Les cibles de RIP dans la signalisation cytotoxique caspase-indépendante des récepteurs de mort ne sont pas encore clairement élucidées. RIP a été imliquée dans la production de ROS dans la signalisation nécrotique du TNFR-1 (Lin et al., 2004b). De plus, la voie de stress impliquant l’activation de JNK (Jun N-terminal Kinase) constitue une des hypothèses. Ainsi, lorsque la caspase-8 est inhibée (par le z-VAD ou par des siRNA spécifiques), RIP1 participe à une mort de type autophagique, via l’activation de JNK (Yu et al., 2004). De plus, en réponse aux dommages à l’ADN, l’activation successive de RIP et de JNK pourrait participer à la perméabilisation des membranes mitochondriales, au relargage d’AIF de la mitochondrie et à l’induction d’une mort de type nécrose-like (Xu et al., 2006). En outre, RIP1 semble impliquée dans l’accumulation de céramide au cours de la mort caspase-indépendante médiée par le TNFα dans plusieurs lignées cellulaires (Thon et al., 2005).
I-7-2 Rôle de la mitochondrie dans la mort caspase-indépendante
La mitochondrie semble jouer un rôle important dans la signalisation caspase-indépendante. En effet, la surexpression de Bcl-2 protège les cellules de la mort de type nécrose-like induite par le TNFα (Thon et al., 2005). De plus, des fibroblastes déficients pour la protéine Apaf1, qui participe à la formation de l’apoptosome, meurent en réponse à la surexpression de tBid, tandis que des fibroblastes doubles déficients pour les protéines Bax et Bak sont résistants. Ces résultats suggérent que les événements mitochondriaux induits par tBid impliquent les membres pro-apoptotiques Bax et Bak et participent à une mort caspase-indépendante (Cheng et al., 2001; Wei et al., 2001). En outre, certaines protéines mitochondriales peuvent être relarguées en l’absence d’activation de caspases. Par exemple, HtrA2/Omi pourrait agir en tant que protéine effectrice dans une mort de type nécrose-like grâce à son activité sérine protéase (Hegde et al., 2002). EndoG est capable d’induire une fragmentation de l’ADN de manière caspase-indépendante sur des noyaux isolés (Li et al., 2001). Enfin, le z-VAD ne prévient pas les effets d’AIF sur la condensation chromatinienne et la fragmentation de l’ADN (Susin et al., 1999).
I-7-3 Les protéases impliquées dans la mort caspase-indépendante
Les autres protéases de l’apoptose, distinctes des caspases, telles que les cathepsines, les calpaïnes et les granzymes (cf I-6) participent aussi aux voies de signalisation caspase-indépendante, notamment en réponse aux agents antitumoraux. La cathepsine B induit la mort non apoptotique de cellules pulmonaires cancéreuses en réponse à des agents stabilisants les microtubules, tel que le paclitaxel (Broker et al., 2004). Les calpaïnes participent à une mort de type apoptose-like induite par la vitamine D dans des cellules MCF-7 du cancer du sein (Mathiasen et al., 2002). Le relargage sélectif d’AIF de la mitochondrie médié par ces deux types de protéases pourrait participer aux voies de signalisation cytotoxique caspase-indépendante (Bidere et al., 2003; Polster et al., 2005). Le granzyme B participe également à l’induction d’une mort caspase-indépendante. En effet, les altérations mitochondriales (relarguage du cytochrome c et chute du potentiel mitochondrial) induites
protéase dans la signalisation caspase-indépendante des récepteurs de mort (Denecker et al., 2001).
I-7-4 Importance de la mort caspase-indépendante en cancérologie
Le succès des traitements anticancéreux repose notamment sur leur capacité à induire la mort des cellules cancéreuses et en particulier par l’induction de l’apoptose dépendante de l’activation des caspases. Cependant, l’activation de voie de signalisation caspase-indépendante semble prometteuse en thérapie anticancéreuse. Ainsi, de nombreux agents chimiothérapeutiques induisent des morts de type apoptose-like ou nécrose-like. Notamment, un panel d’agents antitumoraux incluant l’Ara-C, la Doxorubicine, la Vincristine et le Taxol induisent la mort caspase-indépendante de cellules leucémiques myéloïdes (Carter et al., 2003). La mort de cellules cancéreuses pulmonaires induite par le taxol, un inhibiteur du fuseau mitotique, n’est pas inhibée en présence de z-VAD (Huisman et al., 2002). En réponse à ce même agent, des cellules cancéreuses ovariennes meurent en l’absence d’activation de caspases vraisemblablement par un mécanisme dépendant d’AIF (Ahn et al., 2004).
En outre, l’activation de ces voies alternes de mort cellulaire pourrait permettre de surmonter les résistances des cellules aux traitements. En effet, AIF est impliqué dans la mort caspase-indépendante induite par la staurosporine dans des cellules cancéreuses pulmonaires chimiorésistantes (Gallego et al., 2004). Ainsi, de nouvelles molécules aux propriétés antitumorales induisent la mort de cellules leucémiques U937, de neuroblastomes et de myélomes multiples chimiorésistants, par des mécanismes indépendants des caspases (Ishitsuka et al., 2005; Lee et al., 2006b; Michaelis et al., 2007).
II- La mort des lymphocytes T
La mort cellulaire est un processus essentiel pour la sélection du répertoire lymphocytaire en favorisant notamment l’élimination des clones autoréactifs et des clones qui ne reconnaissent pas les molécules du soi. En périphérie, la mort des lymphocytes activés constitue un mécanisme de rétrocontrôle négatif de la réponse immunitaire. Un défaut de la mort des lymphocytes T participe au développement de maladies auto-immunes comme l’ALPS (Autoimmune lymphoproliferative syndrome).
II-1 Sélection thymique
Les lymphocytes T dérivent de progéniteurs immatures synthétisés dans la moelle osseuse qui migrent ensuite dans le thymus, un organe lymphoïde primaire impliqué dans leur maturation. Après le réarrangement des gènes codant pour les chaînes α et β de leur récepteur d’antigène, le TCR (T-cell receptor), les lymphocytes T immatures en développement dans le thymus subissent une sélection basée sur la spécificité du TCR (Figure 7).
Les thymocytes « double-positifs », c’est-à-dire exprimant à la fois les molécules CD4 et CD8, dont les TCR ne reconnaissent pas les molécules du soi (Complexe Majeur d’Histocompatibilité (CMH)) associées à des peptides antigéniques du soi présentées par les cellules épithéliales du thymus, meurent d’apoptose. A l’inverse, les thymocytes dont le TCR se lie aux molécules du soi surexpriment Bcl-2 et survivent : c’est la sélection positive.
Ensuite, la sélection négative contribue au maintien de la tolérance du soi en permettant l’élimination des lymphocytes potentiellement auto-réactifs. Ainsi, la liaison de trop haute affinité d’un lymphocyte T immature avec des molécules du CMH du soi liées à des peptides du soi exposées à la surface de cellules présentatrices d’antigène, entraîne sa mort. Seuls les thymocytes qui se lient avec une affinité modérée au CMH-peptide du soi survivent et migrent vers les organes lymphoïdes périphériques. Ceux qui reconnaissent le CMH de classe 1 portent alors uniquement le marqueur CD8 (simple positif CD4-CD8+) alors que ceux qui se lient au CMH de classe 2 expriment le marqueur CD4 (simple positif
lpr déficientes pour le récepteur Fas indiquent que la sélection négative dépend de Fas seulement en présence de fortes doses d’antigène tandis qu’elle est indépendante de ce récepteur dans le cas de faibles doses d’antigène (Kishimoto et al., 1998). Cependant, des travaux plus récents in vitro et in vivo à partir de souris lpr et de souris transgéniques exprimant un dominant négatif de la protéine FADD excluent un rôle de Fas dans l’apoptose des thymocytes lors de la sélection thymique (Villunger et al., 2004). Par ailleurs, la surexpression de Bcl-2, ainsi que la déficience en Bim prévient la mort des thymocytes in
vitro et in vivo, suggérant une implication des protéines pro-apoptotiques de la famille de Bcl-2 et en particulier de la protéine Bim dans la sélection négative des thymocytes (Villunger et al., 2004). En outre, il semblerait que des mécanismes de signalisation indépendants des caspases participent à la mort des thymocytes au cours de leur développement puisque l’expression de p35, un inhibiteur des caspases d’origine virale ou le z-VAD (cf chapitre I-3-2 et I-3-3), n’ont pas d’effet sur la sélection négative dans un modèle de souris transgénique (Doerfler et al., 2000).
II-2 Délétion périphérique et AICD (Activation-Induced Cell Death)
La délétion périphérique participe au phénomène de tolérance du soi (par élimination des clones autoréactifs) et constitue un mécanisme de rétrocontrôle négatif de l’activité des lymphocytes (par élimination des lymphocytes T activés) afin d’éviter l’emballement de la réponse immunitaire. Dans les organes lymphoïdes secondaires, les lymphocytes T matures survivent et prolifèrent grâce aux cytokines comme l’IL-2, -4, -6 et -7 qui semblent favoriser l’expression de Bcl-2 et Bcl-xL (Marsden and Strasser, 2003). Les rencontres répétées des lymphocytes T avec les antigènes provoquent l’activation de leur TCR et leur expansion. Les lymphocytes T CD4+ se différencient en cellules T helper de type I (Th1) ou de type 2 (Th2) en fonction de l’environnement cytokinique. Puis, pour maintenir l’homéostasie lymphocytaire, les lymphocytes T activés sont éliminés par AICD en fin de réponse immunitaire. L’IL-2 sensibilise les cellules à la mort en permettant notamment l’augmentation de l’expression de Fas et de FasL à la surface des lymphocytes T ainsi que la diminution de l’expression de FLIP (Budd, 2001). Deux mécanismes ont été proposés concernant l’implication du système Fas/FasL dans l’AICD : (i) une mort par « suicide » dans le cas où le lymphocyte activé exprime à la fois Fas et FasL à sa surface (Brunner et al., 1995; Dhein et al., 1995) ; (ii) une mort « fratricide » entre deux lymphocytes exprimant respectivement le récepteur et le ligand. De plus, il est possible que des cellules non
lymphoïdes exprimant FasL dans les tissus périphériques infiltrés par les lymphocytes T participent à l’AICD (Green et al., 2003). Cependant, l’inhibition de la signalisation de Fas par des anticorps antagonistes de CD95 ou de CD95-L ne prévient pas totalement l’apoptose des lymphocytes T activés induite par un anti-CD3, suggérant l’implication de mécanismes alternatifs indépendants de Fas dans l’AICD (Dhein et al., 1995).
Le rôle du TNF dans l’AICD reste très controversé. En effet, un antagoniste du TNF récepteur I n’a aucun effet sur l’apoptose des lymphocytes T activés induite par l’activation du TCR (Brunner et al., 1995). A l’inverse, une étude récente indique que des anticorps anti-TNF neutralisants inhibent l’AICD (Lawrence and Chow, 2005). Différemment, l’implication de TRAIL dans l’AICD est démontrée dans plusieurs études. Notamment, chez des patients atteints du SIDA (Syndrome d’immunodéficience acquise), la mort des lymphocytes T activés dépend de TRAIL (Katsikis et al., 1997). De même, l’expression de TRAIL augmente au cours de l’activation des lymphocytes T en périphérie et l’inhibition de la signalisation du récepteur par des anticorps antagonistes prévient partiellement l’AICD (Martinez-Lorenzo et al., 1998).
Par ailleurs, certains travaux suggèrent l’implication de mécanismes caspase-indépendants dans l’AICD (Jaattela and Tschopp, 2003). En effet, le z-VAD n’a aucun effet sur la mort de PBL (Peripheral Blood Lymphocytes) activés en réponse à la PHA (Phytohémagglutinine) (Holler et al., 2000). De plus, des lymphocytes T issus de souris portant les mutations lpr et gld sont partiellement résistants à l’AICD et meurent par un mécanisme indépendant du TNFα et des caspases (Davidson et al., 2002). De même, une mort de type nécrotique, indépendante des caspases, est observée dans des lymphocytes T CD4+ et CD8+ après stimulation du TCR. Deux mécanismes distincts ont été proposés : (i) l’un dépendant de Fas et de p38/MAPK (Mitogen Activated Protein Kinase) dans les cellules CD4+ ; (ii) l’autre indépendant de Fas et de p38/MAPK dans les cellules CD4+ et CD8+ (Davidson et al., 2002). En outre, des phénomènes caspases-indépendants tels que le relargage d’AIF et l’activation de JNK ont été décrits au cours de l’AICD de lymphocytes T cytotoxiques, renforçant l’hypothèse de l’existence de voies de signalisation caspase-indépendante dans l’AICD (Chhabra et al., 2006). Finalement, une étude récente suggère un rôle de Granzyme B dans l’AICD de lymphocytes T de type Th2. En effet, à l’inverse des
Enfin, l’ACAD (Activated T Cell Autonomous Death) ou la mort « par négligence » correspond à un second mécanisme de mort des lymphocytes T activés qui dépend de la privation en cytokines. Cette voie de signalisation cytotoxique est inhibée par Bcl-2 et semble impliquer la protéine Bim (Jaattela and Tschopp, 2003).
Figure 7 : Rôle de l’apoptose dans le développement des lymphocytes T (Marsden and Strasser, 2003)
II-3 Pathologies associées à un défaut de la mort des lymphocytes T
Un défaut de mort des lymphocytes T entraîne des maladies du système immunitaire se manifestant par des phénomènes de lymphoprolifération et d’autoimmunité. Des mutations chez la souris et chez l’homme portant sur le récepteur Fas, FasL ou les caspases sont responsables de maladies nommées ALPS (Autoimmune lymphoproliferative syndromes) et rendent comptent de l’importance de ces protéines dans l’apoptose des lymphocytes T (Rieux-Laucat et al., 2003).
Chez l’homme, les ALPS sont causés par une défaillance des mécanismes apoptotiques qui participent au maintien de l’homéostasie lymphocytaire, entraînant l’accumulation de cellules lymphoïdes T double négatives (CD4-CD8-) dans les organes lymphoïdes secondaires et la persistance de cellules autoréactives. L’origine des cellules T double négatives est incertaine ; elles semblent dériver de cellules CD8+ qui ont perdu leur capacité à exprimer CD8, probablement en réponse à une stimulation par un antigène du soi. Ce sont des maladies lymphoprolifératives se manifestant par une lymphadénopathie sévère
accompagnée d'une splénomégalie. De plus, les ALPS sont caractérisées par un syndrome autoimmun se traduisant par une hypergammaglobulinémie et la présence d’auto-anticorps circulants dirigés contre les hématies, les neutrophiles et les plaquettes. Une anémie hémolytique, une neutropénie auto-immune et un purpura thrombopénique auto-immun sont fréquemment retrouvés (Straus et al., 1999).
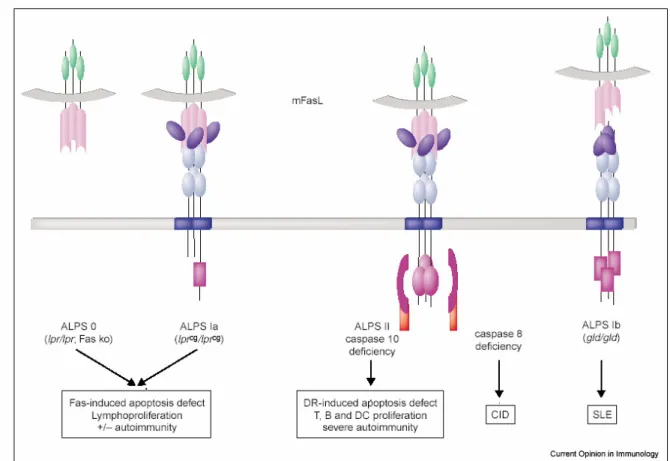
Les différents types d’ALPS avec leurs caractéristiques sont illustrés sur la figure 8. - Dans l’ALPS de type 0, une mutation homozygote de Fas entraîne une déficience complète de la protéine Fas et une forme sévère de la maladie (Rieux-Laucat et al., 1995).
- L’ALPS de type Ia est associé à une mutation hétérozygote sur le gène codant pour Fas, qui induit l’expression d’un récepteur Fas non fonctionnel muté au niveau du domaine de mort. Plus de soixante-dix cas ont été décrits à ce jour. Ces mutations entraînent des défauts d’apoptose par Fas. Il semble que les formes mutées des récepteurs Fas agissent en tant que dominant négatif sur les formes sauvages en perturbant le recrutement de la protéine FADD et la formation du DISC (Martin et al., 1999; Straus et al., 2001).
- Une mutation hétérozygote dominante sur le gène codant pour FasL est responsable de l’ALPS de type Ib chez un patient atteint de lupus érythémateux disséminé. Le phénotype clinique de cette maladie est très différent des ALPS classiques avec une lymphadénopathie mais sans splénomégalie ni accumulation de cellules T doubles négatives. In vitro, les PBLs de ce patient présentent une diminution de l’activité cytotoxique de FasL et sont résistants à l’AICD, tandis que la prolifération des cellules T induite par l’activation du TCR est augmentée (Wu et al., 1996). Par ailleurs, une étude récente décrit un nouveau cas d’ALPS de type Ib dans lequel une forme mutée de FasL exerce un effet dominant négatif sur l’apoptose médiée par Fas. En se liant aux formes sauvages de FasL, il prévient la formation d’un homotrimère biologiquement actif. A l’inverse du cas précédent, le patient présente un phénotype d’ALPS classique (Bi et al., 2007).
- De même, récemment une nouvelle ALPS de type Ic due à une mutation autosomale récessive dans le domaine extracellulaire de FasL a été identifiée chez une patiente présentant les manifestations cliniques et immunologiques d’ALPS sévères (Del-Rey et al., 2006).