HAL Id: tel-02134094
https://tel.archives-ouvertes.fr/tel-02134094
Submitted on 20 May 2019HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Génomique intégrée des tumeurs bénignes
corticosurrénaliennes
Simon Faillot
To cite this version:
Simon Faillot. Génomique intégrée des tumeurs bénignes corticosurrénaliennes. Génétique. Université Sorbonne Paris Cité, 2017. Français. �NNT : 2017USPCB261�. �tel-02134094�
Thèse
Pour obtenir le titre de docteur
Ecole Doctorale : BioSPC
Génomique intégrée des tumeurs
bénignes corticosurrénaliennes
Présentée et soutenue le mardi 28 novembre 2017 par
Simon Faillot
Thèse supervisée par Pr. Guillaume Assié
Composition du Jury :
Rapporteur Rapporteur Examinateur Examinateur Examinateur Dr. Nadia Cherradi Dr. Florence Jaffrezic Dr. Antoine Martinez Pr. Hervé Lefèbvre Dr. Valentina Boeva Université Grenoble-AlpesUniversité Paris-Saclay – INRA Jouy-en-Josas
Université Blaise Pascal - Clermont Ferrand Université de Rouen-Normandie
Remerciements
Je tiens tout d’abord à remercier les docteurs Nadia Cherradi, Florence Jaffrezic,
Valentina Boeva, Antoine Martinez et le professeur Hervé Lefèbvre, qui me font l’honneur
de juger ce travail.
Merci au programme Cancer Research for Personalized Medicine pour avoir financé ma
thèse, me permettant de réaliser mon projet pendant un peu plus de 3 ans.
Je tiens à sincèrement remercier l’ensemble de l’équipe de Pr. Jérôme Bertherat, qui m’a
accueilli depuis début 2014. Un remerciement particulier à mon encadrant Guillaume qui
aura été là jusqu’au bout, y compris dans les périodes les plus difficiles. A Windy, qui
m’aura transmis énormément de ses connaissances et compétences en bioinformatique sur
ma première année au laboratoire, et à qui je dois beaucoup. A « Mama Chiara » et son
mari, qui auront joué un grand rôle dans mon épanouissement actuel, en me présentant
aux bonnes personnes. Merci infiniment à Jérôme Ballif, l’une de ces bonnes personnes.
Merci à Fernande qui m’a beaucoup touché, m’apportant un grand soutien même après
son départ ; ma conjugaison créole étant ce qu’elle est, je ne sais que répondre à « Tiembè
raid » … mais je l’ai fait !
Je tiens également à remercier du fond du cœur Evelyne et Isabelle de m’avoir si bien
accueilli lors de ces derniers mois. Si je suis allé jusqu’au bout, c’est aussi grâce à vous.
Merci à Evelyne et Laurence d’avoir su être à mon écoute.
Merci à l’ensemble de ma famille, qui a su répondre présente en me donnant tout l’amour
dont j’avais besoin (c’est certainement très niais… mais tout aussi vrai !). Merci donc à
Isabelle Pibault (alias « Maman », qui désirait avoir son nom marqué dans ma thèse), à
Jean-Luc Faillot (alias « Papa », parce que pas de jaloux !), à mes frangins adorés Louise
et Marius (et Mojo !), à ma belle-Maman Rita. Mais aussi à mes cousins, oncles et tantes,
qui ont été plus proches que jamais. Merci. Merci aussi à ma deuxième Maman Stéphanie
(ça fait beaucoup de Mamans !), à Chantal, et à leurs enfants, qui sont quelque part aussi
de la famille.
Parlant de famille, merci aux JeCCos (et assimilés), aux JeCCommunicaters, aux
Lobsters… qui sont devenus ma deuxième famille à Cochin : Atsuro (Watashi wa neko
tabemasuuuuuu !), Olivier, Salvi, Stéphane, Marion, Robin, Antonin, RoRo, Mari, Trang,
Armelle, Gaby&Gaby, les espagnols catalans Francesc et « sa coloc » Laura, Nadège,
Gaëlle, Camille, Séverine, Cyril, Loïc, Julien, Maud… tellement de belles rencontres qu’il
est difficile de toutes les énumérer.
Merci à Gaspard et Louis, mes supers amis de toujours, d’avoir été là autant que
vous le pouviez. Merci également à mes colocs, Matthieu, Jordan, Thib, avec qui j’ai
partagé ces dernières années, pour le pire et surtout le meilleur ! Des amis qui m’ont
soutenu dans les moments difficiles (Bérénice, Marguerite, Lucas, Camila, Carino,
l’ensemble de la « team Pollos Hermanos » …). Merci à Luna, le remède à tous les maux.
C’est important, un p’tit chat.
Table des matières
Remerciements ... 1
Table des illustrations ... 5
Liste des abréviations ... 6
Résumé ... 11
I. Introduction ... 12
A. La corticosurrénale et ses tumeurs ... 12
1. La surrénale ... 12 a) Généralités ... 12 b) La médullosurrénale ... 12 c) La corticosurrénale ... 12 2. Le développement de la corticosurrénale ... 14 a) Embryogenèse et organogenèse ... 14
b) Les progéniteurs corticosurrénaliens et leurs marqueurs ... 17
c) Différenciation radiale des cellules avec gradient Wnt/beta-caténine – PKA/AMPc ... 22
3. La stéroïdogenèse corticosurrénalienne ... 24
a) Apport et synthèse du cholestérol, précurseur de la synthèse des hormones stéroïdes . 24 b) Synthèse des hormones stéroïdes à partir du cholestérol ... 26
c) Métabolisme énergétique et détoxification impliqués dans la synthèse de stéroïdes. ... 30
4. Les différentes tumeurs de la corticosurrénale ... 31
a) Les cancers de la corticosurrénale ... 31
b) Les adénomes corticosurrénaliens ... 32
c) Les hyperplasies et dysplasies de la surrénale ... 33
B. Anomalies moléculaires des tumeurs bénignes corticosurrénaliennes et apports de la génomique ... 35
1. Voies de signalisation dérégulées dans les tumeurs surrénaliennes et apports du séquençage haut-débit ... 36
a) L’activation de la voie AMPc/PKA associée aux tumeurs sécrétant du cortisol ... 36
b) L’activation de la voie Wnt/béta-caténine associée aux tumeurs non sécrétantes ... 41
c) Activation de la voie du calcium dans l’hyperaldostéronisme primaire ... 45
d) Anomalies moléculaires liées aux PMAH ... 49
e) Anomalies du cycle cellulaire dans les corticosurrénalomes ... 52
2. Les apports des « Omics » à la connaissance des tumeurs ... 53
a) Le transcriptome ... 53
c) Le méthylome ... 59
d) Les altérations chromosomiques ... 63
e) La génomique intégrée ... 65
II. Résultats ... 66
A. Caractérisation multi-omics des tumeurs bénignes de la corticosurrénale ... 66
1. Article 1 : Génomique intégrée des tumeurs bénignes corticosurrénaliennes... 66
2. Conclusion et perspectives de l’article 1 ... 97
B. Caractérisation de l’exome des tumeurs bénignes de la surrénale ... 98
1. Caractérisation de l’exome des hyperplasies ... 99
a) Echantillons ... 99
b) Méthodes : séquençage/ filtrage ... 99
c) Variants somatiques ... 101
d) Recherche de prédisposition germinales aux hyperplasies ... 102
2. Participation à la caractérisation de l’exome des adénomes de la surrénale ... 103
a) Article 2 : Caractérisation des adénomes corticosurrénaliens non mutés PRKACA ... 104
b) Discussion à propos de l’article 2 ... 117
III. Discussion générale et perspectives ... 118
a) Impact du choix des méthodes bioinformatiques/statistiques ... 118
1. Le clustering : stabilité/méthode ? ... 118
2. Des résultats positifs par le hasard dus à de grands nombres de tests ? ... 121
3. Comment donner un sens biologique à une telle masse de données omics ? ... 122
4. Sensibilité/spécificité du NGS et des méthodes de calling/filtres ... 124
B. Pourquoi s’intéresser aux tumeurs bénignes ? ... 126
C. Les tumeurs bénignes, une anomalie de développement ? ... 128
D. Une stéroïdogenèse anormale, régulée (épi-)génétiquement ? ... 129
E. Comparaison entre tumeurs bénignes et carcinomes, apports éventuels à la compréhension des cancers ... 131
F. Comment allons-nous comprendre les tumeurs sans driver ? ... 132
Bibliographie... 134
Table des illustrations
Figure 1 : La glande surrénale, inspiré de Yates et al. ... 13
Figure 2 : Embryogénèse de la surrénale, d’après Yates et al. ... 15
Figure 3 : Renouvellement du cortex à partir de progéniteurs ... 19
Figure 4: Biosynthèse du cholestérol à partir du squalène ... 25
Figure 5 : La stéroïdogenèse corticosurrénalienne ... 29
Figure 6 : Voie de l’AMPc / Protéine kinase A dans les tumeurs sécrétant du cortisol ... 40
Figure 7 : Voie Wnt / beta-caténine, activée dans les adénomes non sécrétants et l’hyperaldostéronisme primaire ... 44
Figure 8 : Signalisation calcique dans l’hyperaldostéronisme primaire (dépolarisation de la membrane) ... 48
Figure 9 : Comparaison de deux puces à ADN pour l’analyse d’expression, d’après Harrington et al... 55
Figure 10 : ACP de la méthylation des CpG des tumeurs et tissus corticosurrénaliens (d’après Rechache et al) ... 61
Figure 11 : Diagramme de Venn de mutations somatiques par plusieurs analyses de variant calling (d’après Zheng et al, matériel supplémentaire). ... 125
Liste des abréviations
17-OHP : 17α-hydroxyprogestérone
3β-HSD : hydroxy-delta-5-stéroïde déshydrogénase 5-HT : récepteur à la sérotonine (5-hydroxytryptamine) 17β-HSD : 17β-hydroxystéroïde déshydrogénase
βTrCP : Beta-Transducin Repeat Containing E3 Ubiquitin Protein Ligase ACAT : acétylcoenzyme A acétyltransférases ou thiolases
Acétyl-CoA : acétylcoenzyme A
ACP : Analyse en Composantes Principales ACTH : hormone corticotrope
ADNc : ADN complémentaire
AIMAH : ACTH-Independant Macronodular Adrenal Hyperplasia AKR1B1 : aldo-keto reductase family 1 member B1
AMHR : récepteur à l’hormone antimüllérienne AMPc : Adénosine monophosphate cyclique APC : Adenomatous Polyposis Coli
ARMC5 : gène codant armadillo repeat containing protein 5 AT1R : Récepteur à l’angiotensine II de type 1
ATRX : gène codant Alpha Thalassemia/Mental Retardation Syndrome X-Linked ATP : Adénosine triphosphate
ATP1A1 : gène codant la sous-unité α1 de la pompe Na+/K+ ATP2B3 : gène codant la pompe calcique PMCA3
BAF : B-Allele Frequency
BUB1B : gène codant Mitotic checkpoint serine/threonine-protein kinase BUB1 beta C1A / C1B : groupes de corticosurrénalomes de bons et mauvais pronostics
CACNA1D, CACNA1H : gènes codant des canaux calciques voltages-dépendants Cav 1,3 : Calcium channel, voltage-dependent, L type, alpha 1D
CCDC80 : Coiled-Coil Domain Containing 80 CDK4 : gène codant la Cyclin Dependent Kinase 4
CDKN2A : gène codant la protéine Cyclin-dependent kinase Inhibitor 2A CGH : Comparative Genomic Hybridization
CIMP : CpG Island Methylator Phenotype
CpG : dinuclotéide CG : “cytosine-phosphate-guanine” CRE : cAMP-Responsive Element
CREB : cAMP-Responsive Element Binding protein CRH : Cortisol Releasing Hormone
CTNNB1 : gène codant la beta-caténine CYP11A1 : gène codant l’enzyme P450scc
CYP17A1/CYP17 : gène codant l’enzyme 17α-hydroxylase / 17,20 lyase CYP11B1 : gène codant la 11β-hydroxylase
CYP11B2 : gene codant l’aldostérone-synthase CYP19A1 : gène codant l’aromatase
CYP21A2 : gène codant la 21α-hydroxylase
CYP51A1 : gène codant la lanostérol 14α-déméthylase
DAX-1 : dosage-sensitive sex reversale, adrenal hypoplasia critical region on chromosome X, gene 1
DAXX : gène codant death domain associated protein
DHCR7 : gène codant l’enzyme 7-déhydrocholestérol réductase DHEA : déhydroépiandrostérone
DHEA-S : sulfate de DHEA
DICER1 : gène codant une ribonucléase essentielle à la maturation des miRs DNMT : ADN-méthyltransférases
DOC : désoxycorticostérone
ENSAT: European Network for the Study of Adrenal Tumors FAP : Polypose recto-colique familiale
FGD : familial glucocorticoid deficiency FH : gène codant la fumarate hydratase
FRET: Fluorescence Resonance Energy Transfer FOXL2: Forkhead box protein L2
GATK : Genome Analysis Tool Kit GIP: gastric inhibitory peptide GIPR : récepteur au GIP
GIRK4 : G-protein activated Inwardly Rectifying K+ current, canal potassique Gli : Glioma-Associated Oncogene Homolog
GNAS : gène codant la sous-unité α de la protéine Gs Gsa : sous-unité α de la protéine Gs
GSK-3β : glycogène synthase kinase 3 beta
gsp : mutation activatrice R201 de GNAS, à l’origine de l’activité consitutive de Gs GSTP1 : gene codant une Glutathione S-transferase
HDL : High Density Lipoprotein
HMG-CoA : Hydroxyméthylglutaryl-Coenzyme A
HSD3B2 : gène codant la hydroxy-delta-5-stéroïde déshydrogénase IGF-2 : Insulin-llike growth factor 2
INHA : gène codant l’Inhibine a IP3 : inositol triphosphate
IPP : isopentényl-pyrophosphate
KCNJ5 : gène codant pour le canal potassique GIRK4
L205R : mutation de PRKACA, la Leucine 205 du peptide est convertie en Arginine LDL : Low Density Lipoprotein
LDLR : Récepteur aux LDLs
LEF : lymphoid enhancer binding factor LH(R) : (récepteur à l’) hormone lutéinisante LOH : perte d’hétérozygotie
LSS : gène codant l’enzyme lanostérol-synthase MC2R : Melanocortin 2 Receptor, récepteur à l’ACTH miRNA ou miR : micro ARN
MDM2 : Mouse double minute 2 homolog
MM : séquence comportant des erreurs (mismatch)
MS-MLPA : Methylation-specific multiplex ligation-dependent probe amplification NADP+ : nicotinamide adénine dinucléotide
NADPH : forme réduite de la nicotinamide adénine dinucléotide phosphate NGS : Next Generation Sequencing
NR5A1 : gène codant SF-1 NR0B2 : gène codant DAX-1
P450scc : enzyme de clivage de la chaîne latéral du cholestérol (codé par CYP11A1) PAX5, PAX6 : Paired Box 5/6
PINK1 : gène codant PTEN-induced putative kinase 1
PMCA3 : Plasma membrane calcium-transporting ATPase 3 PDE11A, PDE8B : gène codant des phosphodiestérases PIP2 : phosphatidyl inositol diphosphate
PKA : Protein kinase A PLC : phospholipase C
PM : séquence exacte (perfect match)
PMAH : Primary Macronodular Adrenal Hyperplasia
POMC : gène codant le précurseur pro-opiomelanocortin à l’ACTH PPNAD : Primary Pigmented Nodular Adrenal Dysplasia
PRKACA : gène codant la sous unité alpha de la protéine kinase A
PRKAR1A, 2A, 1B, 2B : gènes codant les sous-unités régulatrices de la protéine kinase A PYCARD : gène codant Caspase Recruitment Domain-Containing Protein 5
RB1 : gène du rétinoblastome
RNA-seq : séquençage haut-débit de l’ARN RMA : Robust Multiarray Average
ROS : espèces réactives de l’oxygène
RT-PCR : Real time Polymerase Chain Reaction SCARB1: gène codant SR-B1
SFRP2: Secreted Frizzled-related protein 2
SMAD : Mothers against decapentaplegic homolog, effecteur de la voie TGF-β SR-B1: Scavenger receptor class B member 1
SQLE : gène codant l’enzyme squalène-monooxygénase StAR : steroidogenic acute regulatory protein
SHH : Sonic Hedgehog
SNP: Single Nucleotide Polymorphism TCGA: The Cancer Genome Atlas
TERF2 : gène codant Telomeric repeat-binding factor 2 TERT : gène codant la Telomerase reverse transcriptase TGF-β: Transforming Growth Factor Beta
TGFB2 : Transforming Growth Factor Beta 2 TGFBR1 : Récepteur du TGF-β
TP53 : Tumor Protein p53
TXNRD2 : gène codant les enzymes thiorédoxines réductases Vcf : Variant Calling File
VLDL : Low Density Lipoprotein WGD : Whole-genome doubling WGS : Whole-genome sequencing
WNT : Wingless-type MMTV integration type family WT1 : Wilms tumor 1
zG : zona glomerulosa zF : zona fasciculata zR : zone réticulée
Résumé
Le cortex surrénalien produit des hormones stéroïdes, principalement le cortisol, l’aldostérone et des androgènes. Le cortex surrénalien peut être le siège de tumeurs – adénomes ou cancers-, hyperplasies et dysplasies. Ces lésions sont dans leur grande majorité bénignes. Elles peuvent être associés à une hypersécrétion d’hormone stéroïde, le plus souvent de cortisol (syndrome de Cushing) ou d’aldostérone. Il existe aussi des tumeurs non sécrétantes. Si des classifications moléculaires ont été établies pour les carcinomes, à ce jour il n’existe pas de classification pangénomique des tumeurs bénignes corticosurrénaliennes, qui pourrait renseigner sur les mécanismes de sécrétion autonome et de prolifération de ces lésions. Enfin le déterminisme génétique des dysplasies et hyperplasies n’est que partiellement connu.
Au cours de ma thèse, j’ai pu analyser un jeu de données « omics » complet des lésions bénignes corticosurrénaliennes pour un plus d’une centaine d’échantillons, incluant du séquençage haut-débit (exome/ciblé pour les mutations, RNA-seq pour l’analyse des microARNs), des puces transcriptome et méthylome, et des puces SNP pour la recherche d’altérations chromosomiques. J’ai pu identifier une classification moléculaire pangénomique relativement convergente entre les différentes « omics », qui concorde avec les types tumoraux et sécrétoires, mais identifie également des sous-groupes nouveaux au sein de ces lésions. Il ressort notamment que les mutations dans ces lésions sont des déterminants essentiels de la classification moléculaire. Ainsi sont regroupées les lésions selon la voie de signalisation ou le gène altéré, notamment la voie PKA/AMPc pour les lésions produisant du cortisol, la voie Wnt/beta-caténine pour les adénomes ne sécrétant pas ou peu de cortisol, et ARMC5 pour un sous-groupe d’hyperplasies macronodulaires. Ces groupes très distincts contiennent également des lésions sans mutation identifiée, avec vraisemblablement des mécanismes alternatifs d’altération de ces voies de signalisation.
Dans le groupe des hyperplasies macronodulaires mutées ARMC5, la comparaison avec l’ensemble des autres lésions bénignes fait ressortir une signature d’expression ovarienne forte, marquée par l’expression de FOXL2 et de ses cibles CYP19A1, et PTHLH. Cette marque de différentiation spécifiquement gonadique au sein de la surrénale fait discuter une anomalie de développement.
Cette analyse génomique intégrée identifie également des altérations épigénétiques de la stéroïdogenèse. Notamment les tumeurs sécrétant beaucoup de cortisol sont globalement hyperméthylées dans leurs îlots CpG. Par ailleurs l’hyperméthylation de CYP21A2 est vraisemblablement un mécanisme de déficit en 21-hydroxylase intratumoral. Des signatures de miRNA semblent également avoir un impact sur la stéroïdogenèse.
Au cours de ma thèse j’ai également analysé l’exome des hyperplasies macronodulaires non mutées ARMC5. Je n’ai pas identifié de nouvelle mutation somatique récurrente. Au niveau de l’exome germinal, j’ai identifié plusieurs gènes candidats récurrents, qui ouvrent la voie à des analyses génétiques (extension de cohorte) et de biologie cellulaire complémentaires.
Ce travail est la première caractérisation génomique d’envergure des lésions bénignes de la corticosurrénale. Même si tous les mécanismes ne sont pas élucidés dans le détail, ces données représentent une ressource importante pour orienter les recherches à venir dans la tumorigenèse surrénalienne bénigne et la stéroïdogenèse.
I. Introduction
A. La corticosurrénale et ses tumeurs
1. La surrénale
a) Généralités
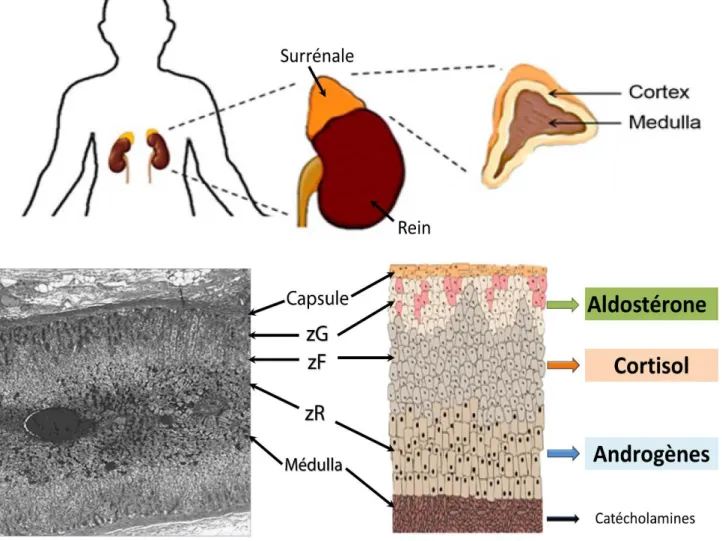
Les surrénales sont deux glandes endocrines situées au dessus des reins. Elles sont composées de la médullosurrénale (au centre) et la corticosurrénale (en périphérie), d’origines embryonnaires et de sécrétions hormonales bien distinctes.
b) La médullosurrénale
La médullosurrénale sécrète des catécholamines, hormones dérivées de l’acide aminé tryptophane : l’adrénaline et la noradrénaline. L’adrénaline, notamment, est sécrétée dans des cellules spécifiques appelées chromaffines. Lors de ma thèse, je me suis uniquement intéressé aux tumeurs de la corticosurrénale.
c) La corticosurrénale
Le cortex surrénalien, ou corticosurrénale, est une glande endocrine caractérisée par sa sécrétion de stéroïdes. Ce cortex présente une zonation tripartite (revue : Vinson, 2016) :
- La zone glomérulée (zG) ou zona glomerulosa, est la zone périphérique. Elle est organisée en paquets de cellules stéroïdogéniques appelées cellules glomérulées.
Ces cellules sécrètent des hormones minéralocorticoïdes, comme l’aldostérone qui participe à la régulation de la tension artérielle en régulant la concentration plasmatique de potassium (kaliémie) ou l’excrétion rénale du sodium.
- La zone fasciculée (zF) ou zona fasciculata, est plus interne. Elle se caractérise par des unités de cellules stéroïdogéniques organisées en cordons radiaux. Ces cellules sécrètent des hormones glucocorticorticoïdes comme le cortisol. Le cortisol a principalement une action hyperglycémiante, mais aussi des actions
sur le métabolisme notamment des graisses. Il agit également sur le système immunitaire avec un rôle anti-inflammatoire.
L’excès de cortisol est associé à plusieurs symptômes, comme des anomalies de répartition des graisses, du diabète, de l’ostéoporose ou de l’hirsutisme. On parle de syndrome de Cushing ; celui-ci peut être dû à des tumeurs corticosurrénaliennes (Newell-Price et al., 2006).
- La zone réticulée (zR), la plus interne et proche de la médullosurrénale, est caractérisée par sa sécrétion d’androgènes, comme la déhydroépiandrostérone (DHEA).
Figure 1 : la glande surrénale, inspiré de Yates et al.
La surrénale est située au dessus des reins. Elle se compose de la médulla au centre et du cortex en périphérie.
Le cortex surrénalien présente une zonation en trois zones concentriques, avec en périphérie la zone glomérulée (zG), de manière intermédiaire la zone fasciculée (zF), et de manière proche de la médulla la zone réticulée (zR).
2. Le développement de la corticosurrénale
a) Embryogenèse et organogenèse
(1)
Développement du primordium adrénogénital à
partir du mésoderme
Le cortex surrénalien, tout comme les gonades, se développe à partir du feuillet intermédiaire de l’embryon ou mésoderme. Dès les premières semaines du développement (4ème semaine) se forme un amas de cellules appelé primordium adrénogénital dans ce qu’on appelle la crête uro-génitale (Yates et al., 2013 pour revue sur l'embryogénèse corticosurrénalienne). Ce primordium donnera naissance aux tissus stéroïdogéniques : le cortex surrénalien, mais également les gonades. Ce primordium est caractérisé par l’expression de Steroidogenic Factor 1 ou SF-1 (Hatano et al., 1996).
(2)
Formation du cortex surrénalien fœtal
Le primordium adrénogénital se divise, donnant naissance à deux primordia distincts, surrénalien et gonadique. Les cellules exprimant le plus SF-1 sont à l’origine du primordium surrénalien (Hanley et al., 1999). L’ensemble du cortex est ensuite entouré d’une fine couche de cellules mésenchymateuses appelée la capsule. A partir du primordium surrénalien se développe la surrénale fœtale, composée d’une capsule, d’un cortex définitif et d’un cortex fœtal, provenant du primordium adrénogénital.
(3)
Développement du cortex définitif
Les cellules du primordium surrénalien continuent de proliférer après individualisation depuis le primordium adrénogénital. Ce primordium est à l’origine d’une surrénale fœtale, qui se vascularise et exprime CYP17 (Hanley et al., 2001). Cette surrénale fœtale produit principalement des androgènes (surtout la déhydroépiandrostérone). Une seconde zone se distingue de cette zone fœtale, le cortex définitif, à l’origine de la surrénale adulte, constitué de petites cellules alors peu sécrétantes. La médulla dérive de cellules issues de la crête neurale, qui migrent tardivement (autour de la sixième semaine de développement chez l’Homme) et forme une couche interne de la surrénale composée de cellules chromaffines.
Cette zonation est claire dès le 2ème mois de développement fœtal chez l’Homme (Xing et al., 2015, autre revue sur le développement). La surrénale fœtale est alors une glande encapsulée, avec au centre des ilots de cellules chromaffines qui formeront plus tard la médullosurrénale, autour de laquelle s’organisent les zones fœtale et définitive du cortex respectivement au centre et en périphérie. Cette surrénale fœtale s’épaissit au cours du développement et jusqu’à la naissance, essentiellement du fait de la croissance de la zone fœtale. Entre 3 et 4 mois de gestation on observe le développement d’une nouvelle zone corticale, intermédiaire aux deux zones précitées : la zone de transition (Mesiano et al., 1993).
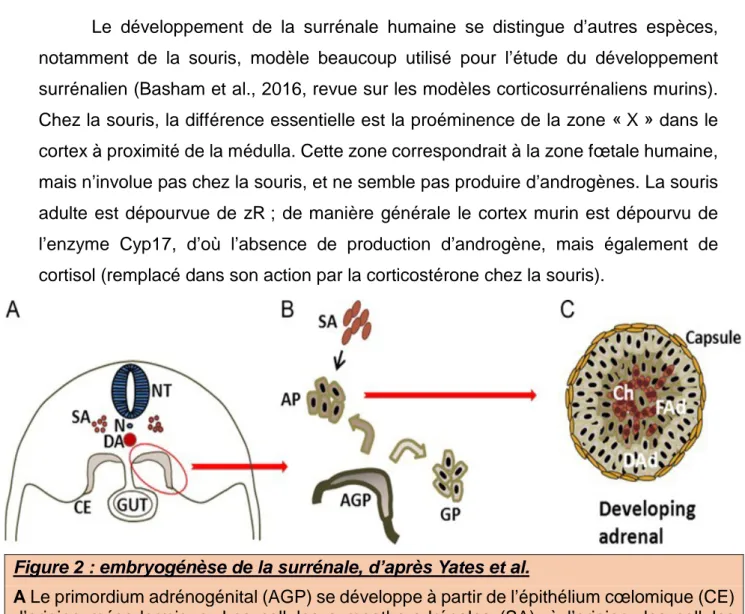
Le développement de la surrénale humaine se distingue d’autres espèces, notamment de la souris, modèle beaucoup utilisé pour l’étude du développement surrénalien (Basham et al., 2016, revue sur les modèles corticosurrénaliens murins). Chez la souris, la différence essentielle est la proéminence de la zone « X » dans le cortex à proximité de la médulla. Cette zone correspondrait à la zone fœtale humaine, mais n’involue pas chez la souris, et ne semble pas produire d’androgènes. La souris adulte est dépourvue de zR ; de manière générale le cortex murin est dépourvu de l’enzyme Cyp17, d’où l’absence de production d’androgène, mais également de cortisol (remplacé dans son action par la corticostérone chez la souris).
Figure 2 : embryogénèse de la surrénale, d’après Yates et al.
A Le primordium adrénogénital (AGP) se développe à partir de l’épithélium cœlomique (CE) d’origine mésodermique. Les cellules sympatho-adrénales (SA), à l’origine des cellules chromaffines, sont initialement adjacentes à la notochorde (N), sous le tube neural (NT). B Les cellules de l’AGP sont marquées par SF-1. Les cellules exprimant le plus SF-1 se séparent de l’AGP pour donner le primordium surrénalien (AP). Les SA migrent ensuite au sein du AP. Les autres cellules de l’AGP donnent naissance au primordium gonadique (GP). C Surrénale en développement, présentant plusieurs zones. Encapsulée, la surrénale fœtale est constituée d’un cortex définitif (DAd) en périphérie, d’un cortex fœtal (FAd) et au centre de cordons de cellules chromaffines (Ch) qui donneront la médulla.
(4) Devenir de la corticosurrénale après la naissance
Chez l’Homme, après la naissance, le cortex définitif se développe et on assiste à la formation des zones glomérulées et fasciculées. Parallèlement, dès les premiers jours suivant la naissance, les cellules de la zone fœtale involuent, et la taille du cortex surrénalien diminue fortement. La production d’androgènes par la surrénale se tarit.A partir de l’âge de 6 ans environ chez l’Homme - et ce jusqu’à la puberté - la ZR devient continue, et s’épaissit ; on observe de nouveau une synthèse d’androgènes. Ce stade est appelé l’adrénarche (Conley et al., 2012).
(5) Steroidogenic factor-1, acteur clef du
développement et de la maturation du cortex
SF-1, codé par NR5A1, est un acteur clef du développement de la corticosurrénale, mais également des gonades (Luo et al., 1994). Il s’agit d’un récepteur orphelin, nécessaire aux développement et la différenciation des tissus stéroïdogéniques, et est un marqueur caractéristique du primordium adrénogénital (Hatano et al., 1996), comme dit précédemment. En effet, des mutations inactivatrices de NR5A1 induisent des dysgénésies des surrénales et/ou des gonades (insuffisances surrénaliennes, ovariennes, stérilité, inversion sexuelle) (Achermann et al., 1999 ; Biason-Lauber and Schoenle, 2000). L’invalidation de Nr5a1 chez la souris engendre une absence de gonades et de corticosurrénale (Luo et al., 1994). Des duplications de ce gène sont très fréquemment associées à des tumeurs pédiatriques de la surrénale, suggérant un rôle dans la tumorigenèse corticosurrénalienne pédiatrique (Figueiredo et al., 2005).
L’expression surrénalienne de 1 est maintenue dans la surrénale adulte. 1 est un acteur majeur du phénotype stéroïdogénique de ces cellules. Notamment SF-1 active directement la transcription des gènes impliqués dans la stéroïdogenèse (Lala et al., 1992 ; Morohashi et al., 1992). Cette spécificité de SF-1 est utilisée en pratique clinique pour affirmer la nature corticosurrénalienne des tumeurs de la surrénale, notamment pour le diagnostic différentiel de métastases surrénaliennes (Sbiera et al., 2010).
L’expression de SF-1 est donc un marqueur important de la différentiation cellulaire surrénalienne, nécessaire au développement mais également au renouvellement des cellules surrénaliennes.
b) Les progéniteurs corticosurrénaliens et
leurs marqueurs
Le cortex surrénalien est une glande qui se renouvelle perpétuellement, avec un renouvellement de ses cellules, mais également une certaine capacité à se régénérer après des lésions, garantissant l’homéostasie du cortex surrénalien. Des expériences sur des rongeurs ont en effet démontré la capacité de la surrénale à se développer de manière compensatoire après une surrénalectomie unilatérale (Israel et al., 1986). D’autres expériences dites d’énucléation surrénalienne - consistant au retrait de la médulla et du cortex pour ne laisser que la capsule et les parties les plus externes de la zG - ont montré la capacité du cortex à se régénérer (Ingle and Higgins, 1938).
Cette capacité de régénération implique que la surrénale contient, en plus des cellules stéroïdogéniques différenciées, des cellules indifférenciées / peu différenciées - proches de cellules souches - capables de se diviser et de se différencier pour produire l’ensemble des cellules du cortex surrénalien. Il s’agit d’une population hétérogène de cellules, présentes en périphérie du cortex, au niveau de la capsule et dans les parties les plus externes de la zG, montrées par des expériences de marquages (King et al., 2009).
La connaissance de ces mécanismes n’est aujourd’hui pas exhaustive, mais des travaux sur des rongeurs ont apporté beaucoup d’éléments pour la connaissance des lignées de cellules à l’origine du développement et du renouvellement surrénalien. Plusieurs populations de cellules progénitrices ou potentiellement progénitrices ont été identifiées.
(1) Progéniteurs sous-capsulaires, avec activation de
Sonic Hedgehog (SHH+)
Une lignée de progéniteurs, située sous la capsule dans la zone glomérulée, exprime le marqueur Sonic Hedgehog, ou SHH (King et al., 2009). Ces progéniteurs
ont deux origines : (i) une population est issue du cortex fœtal et du primordium surrénalien, exprimant SF-1 ; (ii) une seconde population de progéniteurs n’exprime pas Sf1 (SHH+/SF1-), et semblent donc ne pas avoir de différenciation stéroïdogénique. Cependant ces cellules sont capables de devenir stéroïdogéniques (SHH+/SF1+). Les descendants de ces deux types de progéniteurs migrent de manière radiale et centripète, et se différencient afin de donner naissance à l’ensemble des cellules stéroïdogéniques du cortex surrénalien.
(2) Progéniteurs capsulaires GLI+
Une seconde population non-stéroïdogénique peu différenciée se situe principalement dans la capsule surrénalienne, et occasionnellement sous la capsule. Ces cellules expriment GLI1, cible de la voie SHH (GLI+/SHH-) (King et al., 2009). Ces cellules semblent contribuer au développement et au maintien de la capsule du cortex. Notamment ces cellules peuvent se différencier en cellules progénitrices SHH+, sous l’induction paracrine de SHH produits par des cellules sous capsulaires.
(3) Cellules WT1+
WT1 (Wilms tumor 1) est normalement un marqueur du primordium adrénogénital, qui s’éteint dans la surrénale fœtale (Vidal and Schedl, 2000). WT1 est - tout comme SF-1 qui en est une cible (Val et al., 2007) - nécessaire au développement du primordium adrénogénital (Moore et al., 1999). WT1 semble responsable du maintien des cellules primordiales adrénogénitales dans un état indifférencié ; sa répression est en effet nécessaire à la différenciation des cellules en cellules stéroïdogéniques surrénaliennes (Bandiera et al., 2013). GLI1 est une cible de WT1 (Bandiera et al., 2013).
Malgré « l’extinction » de WT1 au stade de surrénale fœtale, des cellules Wt1+ sont détectées dans le cortex de souris adultes (Bandiera et al., 2013). Il existe plusieurs populations de cellules Wt1+, certaines Wt1+/Gli+, d’autres Wt1+/Gli-. La localisation de ces cellules est observée dans la capsule, mais aussi dans le cortex sous capsulaire. Le lien éventuel entre ces cellules WT1+ et les progéniteurs SHH+ et GLI+ décrits précédemment n’est pas complètement clarifié à ce jour. Bandiera et ses collègues montrent que ces cellules WT1+ sont capables de se différencier en cellules corticosurrénaliennes.
Figure 3 : Renouvellement du cortex à partir de progéniteurs
La capsule et la zone sous capsulaire présentent des populations de cellules progénitrices, qui se divisent et donnent naissance au tissu corticosurrénalien. Les cellules migrent de manière centripète, se différencient en cellules de la zG puis de la zF avant de mourir au niveau de la zR. Certains progéniteurs sous la capsule expriment SHH, et peuvent ou non exprimer SF-1. Une autre population exprime GLI, dans la capsule ou sous la capsule. Les descendants des progéniteurs GLI+ peuvent également donner naissance aux cellules de la capsule. Sous l’induction de Sonic Hedgehog, ces progéniteurs peuvent eux-mêmes exprimer SHH. Enfin, des progéniteurs WT1 provenant du primordium adrénogénital existent, et peuvent également donner naissance au tissu stéroïdogénique du cortex.
(4)
Maintien du caractère pluripotent des
progéniteurs capsulaires et sous-capsulaires via DAX-1
Le développement et le renouvellement du cortex surrénalien fait donc intervenir des populations de progéniteurs dans et sous la capsule. Afin de pouvoir se diviser et donner naissance à l’ensemble du tissu surrénalien, ces cellules doivent être maintenues dans un état indifférencié.Le maintien de cette pluripotence fait notamment intervenir le récepteur nucléaire orphelin dosage-sensitive sex reversal, adrenal hypoplasia critical region on chromosome X, gene 1 (DAX-1), codé par NR0B2. DAX-1 a été identifié comme une cause d’hypoplasie congénitale des surrénales liée à l'X. Des mutations ou délétions de ce gène provoquent un hypogonadisme et une insuffisance surrénalienne, avec un développement anormal du cortex surrénalien (Muscatelli et al., 1994 ; Zanaria et al., 1994).
L’invalidation expérimentale de Dax-1 sur des modèles murins engendre une différenciation excessive et prématurée des progéniteurs capsulaires et sous-capsulaires. Cette différenciation induit la perte de la population de progéniteurs (Scheys et al., 2011). DAX-1 exerce un rôle de répresseur sur SF-1, en inhibant la transcription des gènes cibles de SF-1 impliqués dans la stéroïdogenèse (Crawford et al., 1998 ; Ito et al., 1997). DAX-1 est donc un régulateur essentiel du maintien des cellules progénitrices corticosurrénaliennes dans un état indifférencié.
(5)
Tumorigenèse surrénalienne murine après
gonadectomie : une démonstration de cellules
stéroïdogéniques à potentiel gonadique dans les
surrénales
Des expériences sur des modèles murins ont montré que des souris gonadectomisées développent des tumeurs surrénaliennes (Woolley et al., 1943). Ce développement implique des cellules pluripotentes, localisées au sein de la capsule entourant le cortex surrénalien, et qui sont Wt1+/Gli+. Ces progéniteurs fœtaux donnent naissance à des cellules stéroïdogéniques très particulières, caractérisées par des marqueurs gonadiques au sein du cortex, comme le montre l’expression du récepteur à l’hormone lutéinisante Lhr, de Cyp17 - qui ne s’exprime que dans les
gonades chez la souris-, du facteur de transcription Gata4, du récepteur de l’hormone antimüllérienne Amhr, et du facteur de transcription Foxl2 (Bandiera et al., 2013 ; Dörner et al., 2017).
FOXL2 (Forkhead box protein L2), notamment est un facteur de transcription essentiel du développement ovarien. Il active notamment CYP19A1, codant l’aromatase à l’origine de la synthèse des œstrogènes dans l’ovaire (Batista et al., 2007 ; Pannetier et al., 2006 ; Uhlenhaut et al., 2009). Des mutations activatrices de
FOXL2 sont fréquemment décrites dans des tumeurs ovariennes (Shah et al., 2009).
D’autres mutations, inactivatrices sont causes d’insuffisance ovarienne prématurée (Crisponi et al., 2001), Des expériences de knock-out de Foxl2 chez la souris ont également montré un défaut de développement ou de maintien de l’ovaire (Schmidt et al., 2004 ; Uda et al., 2004). FOXL2 est normalement faiblement détecté dans le cortex surrénalien, et est principalement fortement exprimé dans l’ovaire (Yang et al., 2010). Il a également été montré comme interagissant avec SF-1 et le récepteur aux mélanocortines de type 2 (MC2R) (Jin et al., 2016 ; Park et al., 2010 ; Yang et al., 2010), et un rôle dans l’inhibition de STAR (Steroidogenic acute regulatory protein) a également été décrit (Pisarska et al., 2004). Enfin, une publication récente décrit que l’action de FOXL2 dans la différenciation des cellules ovariennes semble induite par la beta-caténine (Li et al., 2017).
Le modèle de la souris gonadectomisée repose sur deux mécanismes de dérégulations hormonales. (I) Une augmentation de l’hormone lutéinisante circulante (LH), du fait du découplage entre les ovaires et le complexe hypothalamo- hypophysaire. Les cellules du cortex surrénalien expriment alors de manière ectopique LHR (Bernichtein et al., 2009). (II) Une diminution de l’inhibine circulante, en provenance des gonades.
Des modèles de souris invalidées Inha (Inhibine a) ont été développés. La quasi-totalité des souris développent très tôt des tumeurs surrénaliennes, souvent de manière bilatérale (Matzuk et al., 1994). Cela donne un premier aperçu du rôle de la voie des TGF-β et des inhibines dans la surrénale.
L’inhibine interfère avec la signalisation du Transforming Growth Factor Beta, par une action antagoniste de l’activation des SMAD, notamment SMAD3 (Mothers against decapentaplegic homolog 3). Les SMAD sont les effecteurs du TGF-β, et sont
activés par phosphorylation. Dans la périphérie des cortex surrénaliens de souris invalidés Inha, couplé à une gonadectomie, une accumulation de SMAD3 phosphorylé est en effet observé (Beuschlein et al., 2003). A l’inverse, un knock-out de Smad3 freine la tumorigenèse surrénalienne des souris gonadectomisées invalidées Inha. Cela démontre le rôle de SMAD3 et de la voie des TGF-β dans ces tumeurs (Looyenga and Hammer, 2007). L’hormone lutéinisante participe à cette activation : son induction permet l’expression du récepteur aux TGF-β, TGFBR1, ainsi que TGFB2, son ligand et activateur de la voie. Finalement, l’action de SMAD3 après activation par la voie TGF-β engendre l’expansion de cellules qui expriment de nouveau le facteur de transcription GATA4, marqueur du développement des cellules gonadiques (Heikinheimo et al., 1997).
c) Différenciation radiale des cellules avec
gradient Wnt/beta-caténine – PKA/AMPc
(1)
Migration et différenciation des cellules
Des expériences in vitro ont montré que les cellules du cortex migrent de manière centripète depuis la zone glomérulée jusqu’à la zone fasciculée, puis la zone réticulée (Freedman et al., 2013).
Le suivi de cellules a montré non seulement que des cellules migrent de la zG vers la zF, mais qu’en plus les cellules de la zG -sécrétant de l’aldostérone- évoluent en cellules de la zF, sécrétant du cortisol. En effet, des expériences suivant des cellules sur modèles murins montre que des mêmes cellules expriment d’abord
CYP11B2 - codant l’aldostérone synthase, spécifique de la zG - avant de surexprimer CYP11B1 - codant la 11β-hydroxylase, spécifique de la zF (Freedman et al., 2013).
C’est donc non seulement la démonstration d’une migration radiale des cellules différenciées stéroïdogéniques au sein du cortex surrénalien, mais également celle d’une différenciation de cellules spécialisées sans passer par une dédifférenciation, de cellules différenciées de la zG produisant des minéralocorticoïdes vers les cellules différenciées de la zF sécrétant des glucocorticoïdes.
(2)
Homéostasie de la zonation zG/zF
La zone glomérulée, ainsi que certains progéniteurs (SHH+) sont inductibles par WNT4 (Walczak et al., 2014), qui est critique pour le développement de la zone glomérulée. WNT4, ainsi que l’aldostérone synthase (Ogishima et al., 1992) et le récepteur à l’angiotensine AT1R (Frei et al., 2001 ; Harada et al., 2010), sont des marqueurs de la zone glomérulée ; ils ne sont en effet pas exprimés dans la zF.
Les cellules de la zG expriment le récepteur à l’ACTH, MC2R (Gorrigan et al., 2011), et sont caractérisées par l’activité de la voie de signalisation AMPc/PKA. L’activation de cette voie, notamment via MC2R, engendre l’orientation de cellules de la zG vers une différenciation en cellules de la zF (Chida et al., 2007). Le maintien et l’homéostasie de la zG repose par une inhibition de la voie de l’AMPc, effectuée par la voie Wnt/beta-caténine via CCDC80 (Walczak et al., 2014). CCDC80 serait une protéine secrétée sous la régulation de la voie Wnt, qui inhiberait la différenciation de la zG en cellules de la zone fasciculée, mais également impliquée dans le maintien des progéniteurs de la zG dans un état indifférencié (Walczak et al., 2014).
Inversement, la différenciation en cellules de la zF nécessite l’inhibition de la voie Wnt/beta-caténine. Celle-ci est effectuée par la protéine kinase A (Drelon et al., 2016) : l’activation de la voie de l’AMPc est responsable de l’inhibition de la voie Wnt, et induit la différenciation des cellules de la zG vers des cellules de la zF.
Ces deux voies de signalisation, Wnt/beta-caténine et AMPc/PKA, sont ainsi nécessaires à l’homéostasie des zones fasciculées et glomérulées. Elles sont toutes les deux cruciales dans la prolifération et la différenciation des cellules corticales et sont impliquées dans la tumorigenèse bénigne. Je reviendrai plus tard sur cette signalisation lorsque j’aborderai les anomalies moléculaires des tumeurs bénignes corticosurrénaliennes.
3. La stéroïdogenèse corticosurrénalienne
a) Apport et synthèse du cholestérol,
précurseur de la synthèse des hormones
stéroïdes
La synthèse des stéroïdes dans le cortex surrénalien se fait à partir du cholestérol. L’apport du cholestérol se fait par l’alimentation ; jusqu’à 80% du cholestérol surrénalien à l’origine de la stéroïdogenèse provient de cholestérol circulant (Borkowski et al., 1967) .
Le cholestérol étant hydrophobe, son transport par le sang jusqu’aux surrénales se fait par le biais de lipoprotéines (HDL, LDL ou VLDL, selon la densité des lipoprotéines), sous forme d’esters de cholestérol. L’entrée des esters de cholestérol dans la surrénale est facilité par des récepteurs membranaires. Les LDLs sont principalement pris en charge par les LDLR (récepteur aux lipoprotéines de basse densité). La prise en charge des HDLs est effectuée par SR-B1 (codé par SCARB1 pour Scavenger receptor class B member 1) (Acton et al., 1996). Les deux récepteurs sont impliqués dans l’absorption d’esters de cholestérol par la cellule chez l’Homme, chez la souris c’est essentiellement le rôle de SR-B1, dont l’expression est notamment induite par l’ACTH et l’angiotensine II (Liu et al., 1997 ; Rigotti et al., 1996 ; Cherradi et al., 2001). Les esters de cholestérol sont stockés au niveau de gouttelettes lipidiques. Le cholestérol est rendu disponible pour la cellule via une hydrolyse des esters de cholestérol, effectuée par les lipases.
Une synthèse de novo endogène du cholestérol existe également dans la surrénale. Pour preuve, le gène DHCR7 - codant la 7-déhydrocholestérol réductase, enzyme responsable de la conversion de 7-déhydrocholestérol en cholestérol - peut notamment impliquer une insuffisance surrénalienne lorsqu’il est défectueux (Nowaczyk et al., 2001). Elle fait intervenir la voie du mévalonate, à partir de l’acétyl-CoA (voir Miziorko, 2011 pour revue).
Cette synthèse débute au niveau du cytosol, où plusieurs acétyl-CoA sont condensés en hydroxyméthylglutaryl-CoA (HMG-CoA). Cette condensation fait intervenir plusieurs enzymes, que sont les thiolases (Acetyl-CoA Acetyltransférases ou ACAT) et la HMG-CoA synthase. Le HMG-CoA est réduit en mévalonate.
Plusieurs réactions se suivent, résultant de la formation de petits hydrocarbures insaturés pyrophosphatés à 5 carbones, dont l’IPP (isopentényl-pyrophosphate). Ces molécules sont précurseurs du motif isoprène, retrouvé de manière générale dans les terpènes et ainsi dans les stéroïdes.
A l’issue de la voie du mévalonate est synthétisé le squalène, hydrocarbure insaturé à 30 carbones, précurseur de tous les stérols et donc des hormones stéroïdes. Le squalène est rendu cyclique pour former le lanostérol, faisant intervenir les enzymes squalène-monooxygénase et lanostérol-synthase, codées respectivement par SQLE et LSS. De nombreuses réactions sont ensuite nécessaires pour convertir la lanostérol en cholestérol, selon des mécanismes complexes faisant notamment intervenir dans sa première étape la lanostérol 14α-déméthylase, codée par CYP51A1 (cf figure 4, ci-dessous). La conversion de déhydrocholestérol en cholestérol par la 7-déhydrocholésterol réductase est l’étape finale de cette synthèse.
Figure 4: Biosynthèse du cholestérol à partir du squalène (Marijanovic et al., 2003)
Abréviations : SQLE : Squalene monooxygenase, LSS : Oxidosqualene cyclase, TM7SF2 : Stérol
Δ14-oxidase, CYP51 : lanostérol 14α-déméthylase, SC4MOL : Stérol Δ4-methyl Δ14-oxidase, H105E3/NSDHL : 3Δ -Hydroxysteroid dehydrogenase, HSD17B7 : 3-Ketostéroïde reductase, EBP : 3Δ-Hydroxystéroïde Δ8-Δ7-isomérase, SC5D : 3Δ-Hydroxystéroïde Δ5-désaturase, DHCR24 : 3Δ-Hydroxysteroid Δ24-reductase, DHCR7 : 7-dehydrocholesterol réductase
b) Synthèse des hormones stéroïdes à partir du
cholestérol
(1)
Clivage de la chaîne latérale du cholestérol, étape
limitante de la stéroïdogenèse
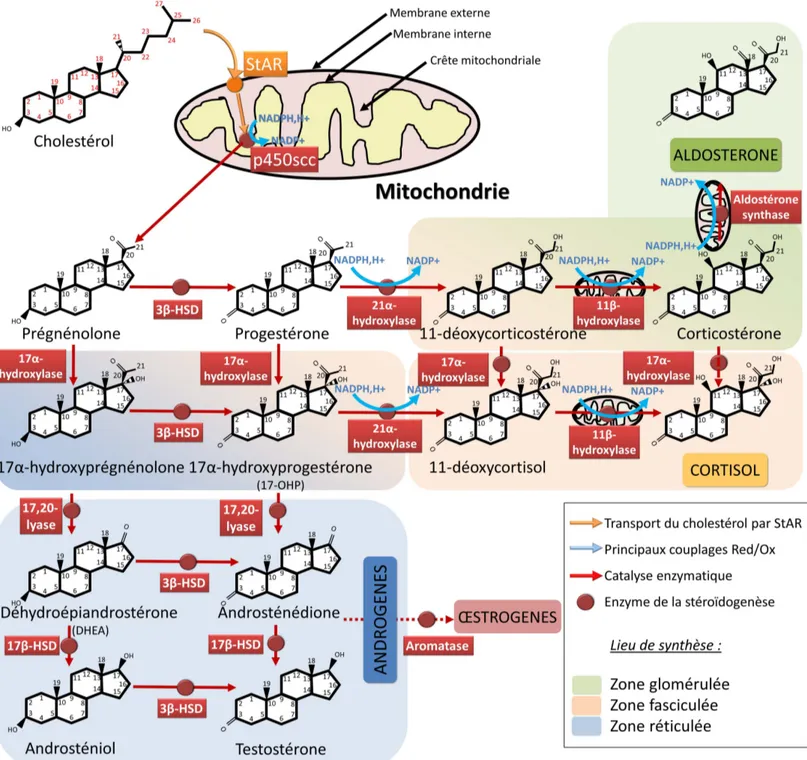
La première étape clef de la synthèse du cholestérol est le clivage de la chaîne latérale du cholestérol par la cholestérol monooxygénase, l’enzyme de clivage de la chaîne latérale du cholestérol (P450scc), codée par CYP11A1 (Miller and Auchus, 2011 pour revue). Sa principale action est de convertir le cholestérol en prégnénolone, stéroïde à 21 carbones (comme l’aldostérone et le cortisol). La prégnénolone est précurseur de la synthèse de l’ensemble des hormones stéroïdes, cette réaction est donc clef de la stéroïdogenèse (Stone and Hechter, 1954).
Cette enzyme est une protéine mitochondriale et son action se fait dans la membrane interne de la mitochondrie (Yago and Ichii, 1969). Du fait de la nature aqueuse de l’espace intermembranaire mitochondrial et du caractère hydrophobe du cholestérol, le transport du cholestérol dans la mitochondrie se fait via le transporteur StAR (pour Steroidogenic Acute Regulatory protein, codée par STAR) (Jefcoate et al., 1987 ; Stevens et al., 1984). CYP11A1 et STAR sont tous deux acteurs clefs de la stéroïdogenèse ; un défaut de l’un de ces deux gènes est la cause d’hyperplasies congénitales lipoïdes des surrénales, une maladie sévère causant l’absence de synthèse stéroïdienne aussi bien surrénalienne que gonadique, et souvent léthale (Lin et al., 1995).
Cette réaction fait intervenir des réactions d’oxydoréduction, où plusieurs NADPH sont oxydés en NADP+, de l’oxygène est réduit en eau. Ces réactions font intervenir deux autres enzymes que sont la ferrédoxine et l’adrénodoxine réductase, qui forment avec P450scc le complexe de clivage de la chaîne latérale de cholestérol, et couplent le transfert d’électron du NADPH vers P450scc. Ce couplage n’est pas exhaustif et il en résulte la production d’espèces réactives de l’oxygène, néfastes pour la cellule (Li et al., 1995).
(2)
Synthèse du cortisol et de l’aldostérone
Les deux principales hormones stéroïdiennes surrénaliennes chez l’Homme, le cortisol et l’aldostérone, sont toutes deux issues du précurseur prégnénolone (Stone and Hechter, 1954).
Leurs voies de synthèse sont similaires (cf. figure sur la stéroïdogenèse surrénalienne). La principale différence entre ces deux voies réside en une hydroxylation au niveau du carbone 17 dans la synthèse des glucocorticoïdes. Cette hydroxylation est effectuée par la 17α-hydroxylase, codée par CYP17A1. Elle s’effectue principalement à partir de deux substrats : la prégnénolone, convertie en 17-hydroxyprégnénolone, ou la progestérone, convertie en 17α-hydroxyprogestérone (17-OHP).
Pour les glucorticoïdes, la 17-hydroxyprégnénolone est convertie en 17-OHP par l’oxydation du groupement hydroxyle en C3 en cétone. Cette oxydation se fait via la hydroxy-delta-5-stéroïde déshydrogénase (3β-HSD) codée par HSD3B2. Le carbone 21 de la 17-OHP est hydroxylé par la 21α-hydroxylase - codée par CYP21A2 - pour former le précurseur 11-déoxycortisol. Ces deux étapes ont lieu au niveau du réticulum endoplasmique.
La 11β-hydroxylase, codée par CYP11B1, hydroxyle le carbone C11 du 11-déoxycortisol pour donner le cortisol. Cette étape est similaire à celle réalisée par l’enzyme de clivage de la chaîne latérale du cholestérol. Elle a en effet lieu dans la membrane interne mitochondriale, et met également en jeux des transferts d’électrons similaires (Yago and Ichii, 1969). Elle engendre donc également une production de radicaux libres dans la mitochondrie.
En ce qui concerne les minéralocorticoïdes, similairement à la synthèse du cortisol, la prégnénolone est convertie en progéstérone par la 3β-HSD, elle-même hydroxylée en désoxycorticostérone (DOC) par la 21 hydroxylase.
Enfin, la DOC est convertie en corticostérone puis en aldostérone par l’aldostérone-synthase (codée par CYP11B2). Cette enzyme est exprimée spécifiquement au niveau de la zone glomérulée. La conversion en corticostérone résulte d’une 11β-hydroxylation de la DOC. La corticostérone est le principal glucocorticoïde chez la souris, dont le cortex est dépourvu de 17α-hydroxylase. La
corticostérone est hydroxylée en C18 par cette enzyme, puis ce groupement hydroxyle est oxydé en cétone. Cette conversion a également lieu dans la mitochondrie.
(3)
Synthèse d’androgènes dans la zone réticulée
Le cortex surrénalien est également lieu d’une synthèse d’androgènes, qui n’est donc pas exclusive aux testicules. Celle synthèse a lieu dans la zone réticulée, où sont produits plusieurs hormones, dont le déhydroépiandrostérone (DHEA).
Contrairement aux dérivés de la progestérone comme le cortisol qui comptent 21 carbones, on dénombre 19 carbones pour les androgènes. Cette perte de carbone se fait via la 17α-hydroxylase, qui possède également une activité 17,20 lyase. Elle clive la chaîne latérale des stéroïdes, laissant place à une fonction cétone au niveau du carbone C17. La 17,20 lyase a deux substrats, la 17α-hydroxyprégnenolone et la 17-OHP, respectivement converties en DHEA et en androsténédione (cf. figure sur la stéroïdogenèse).
Les conversions respectives de la DHEA en androsténédiol, et de l’androsténédione en testostérone, nécessitent une 17β-hydroxystéroïde déshydrogénase (ou 17β-HSD), dont il existe plusieurs isoformes. La principale isoforme de la zone réticulée est HSD17B5, codée par AKR1C3. La réaction catalysée est une réduction de la fonction cétone en C17.
La DHEA peut également être sulfatée en C3 par SULT2A1 pour donner l’androstérone sulfate (ou DHEA-S).
L’androsténédione et la testostérone sont précurseurs de la synthèse des œstrogènes via l’aromatase codée par CYP19A1. Cette synthèse n’a normalement pas lieu dans le cortex surrénalien - a priori dépourvu d’aromatase - mais dans les ovaires.
Figure 5 : La stéroïdogenèse corticosurrénalienne
Les principales étapes de la stéroïdogenèse sont ici représentées, avec le lieu de synthèse privilégié au sein du cortex surrénalien pour chaque stéroïde. La synthèse des œstrogènes n’a pas lieu dans la surrénale, qui est normalement dépourvue d’aromatase.
Abréviations : P450scc : cytochrome P450 side-chain clivage (enzyme de clivage de la chaîne latérale du cholestérol, codée par CYP11A1) ; HSD : 3/17β-hydroxystéroïde déshydrogénase.
c) Métabolisme énergétique et détoxification
impliqués dans la synthèse de stéroïdes.
Comme décrit précédemment, la synthèse de stéroïdes nécessite des couplages redox avec des enzymes de la stéroïdogenèse. Cela implique le couple NADP+/NADPH dans ces réactions d’oxydoréduction.
Il est nécessaire pour la cellule de maintenir un équilibre entre NADP et NADPH. Notamment dans la mitochondrie, où les enzymes stéroïdogéniques (l’enzyme de clivage de la chaîne latérale du cholestérol, la 11β-hydroxylase et l’aldostérone synthase) oxydent toutes le NADPH en NADP+. En outre, les couplages imparfaits avec les enzymes entraînent un transfert d’électrons causant la formation de radicaux libres, comme l’ion superoxyde. Une détoxification au sein de la cellule est alors nécessaire pour éviter un stress oxydant.
Des mutations du gène NNT, codant la Nicotinamide Nucléotide Transhydrogénase, a été identifiée dans des cas de syndrome de déficit isolé en glucocorticoïdes (ou FGD pour familial glucocorticoid deficiency) (Meimaridou et al., 2012). Cette protéine permet la réduction de NADP+, afin de rétablir une concentration élevée en NADPH mitochondrial, nécessaire dans des étapes clefs de la stéroïdogenèse corticosurrénalienne.
Une teneur élevée en NADPH mitochondrial permet de maintenir un ratio de glutathions réduits élevés. Les glutathions sont la principale source de détoxification des espèces réactives de l’oxygène (ROS) dans la cellule.
L’autre système de détoxification des ROS mis en jeu est celui des thiorédoxines. Ce sont des protéines oxydantes qui interviennent dans la réduction des ROS par formation de pont disulfures. Les thiorédoxines sont maintenues réduites par les enzymes thiorédoxines réductases, codées notamment par TXNRD2. Des mutations de ce gène ont été décrites dans des cas de FGD, mettant une nouvelle fois en évidence l’importance de la régulation du stress oxydant dans les mitochondries dans la stéroïdogenèse (Prasad et al., 2014).
D’autres produits secondaires de la stéroïdogenèse, toxiques pour la cellule, doivent être éliminés. C’est le cas de l’isocaproaldéhyde (4-méthylpentanal), issu du clivage de la chaîne latérale du cholestérol (Constantopoulos et al., 1966). Cet
aldéhyde est notamment dégradé en alcool, via une réduction réalisée par des aldo-cétones réductases (Lefrançois-Martinez et al., 1999). Akr1b7 est responsable de cette réduction chez la souris ; chez l’Homme AKR1B1 réalise une réaction similaire (Pastel et al., 2016).
4. Les différentes tumeurs de la
corticosurrénale
Le cortex surrénalien peut être le siège de tumeurs. Elles peuvent être malignes (carcinomes corticosurrénaliens ou corticosurrénalomes) ou bénignes, peuvent être associées ou non à des prédispositions héréditaires (comme dans le cas des hyperplasies congénitales des surrénales), des raretés diverses et des types de sécrétions hormonales différentes.
Au cours de ma thèse, mon travail a été restreint aux tumeurs bénignes de la corticosurrénale, les adénomes et hyperplasies. Je n’ai pas travaillé sur les hyperplasies congénitales des surrénales, dont la génétique et les causes ont déjà été extensivement décrites.
a)
Les cancers de la corticosurrénale
Les carcinomes surrénaliens, ou corticosurrénalomes, sont des tumeurs agressives rares (prévalence estimée entre 0.5 et 2 cas par million et par an chez l’adulte) se développant à partir du cortex surrénalien (Golden et al., 2009 ; Kerkhofs et al., 2013). Elles sont associées à un pronostic sombre, avec une survie à 5 ans inférieure à 40% (Fassnacht et al., 2009 ; Icard et al., 2001 ; Lughezzani et al., 2010). Cependant l’agressivité du carcinome varie beaucoup en fonction du patient ; on distingue en effet des cancers très agressifs, ayant tendance à récidiver et à développer des métastases, et des tumeurs indolentes, pouvant être guéries par la chirurgie.
Même si des thérapies existent (mitotane, chimiothérapie), la thérapie la plus efficace reste le retrait chirurgical (Allolio et al., 2004).
Ces tumeurs malignes, même si étudiées au laboratoire, n’ont pas fait l’objet de mes recherches lors de ma thèse.
b) Les adénomes corticosurrénaliens
(1)
Les adénomes de Conn
Les adénomes produisant de l’aldostérone - ou adénomes de Conn - sont associés à une élévation de l’expression de l’aldostérone synthase, et se développent au dépend de la zone glomérulée du cortex surrénalien (Conn, 1955). On parle d’hyperaldostéronisme primaire, car l’excès de production d’aldostérone n’est pas dû au contrôle du système rénine-angiotensine. On estime que l’hyperaldostéronisme primaire représente environ 10% des cas d’hypertension (Monticone et al., 2017), même si cette prévalence varie en fonction de la population étudiée (Karashima et al., 2017). Les adénomes de Conn comptent pour le tiers des cas d’hyperaldostéronisme primaire, les autres cas étant divers types d’hyperplasies du cortex surrénalien, sur lesquelles je ne me suis pas focalisé pendant ma thèse.
(2)
Les adénomes cortisoliques
Ces tumeurs sont à l’origine de syndrome de Cushing indépendant de l’ACTH hypophysaire. On estime en effet qu’environ 10% des cas de syndrome de Cushing sont dus à un adénome surrénalien (Newell-Price et al., 2006). L’adénome se présente le plus souvent sous la forme d’un unique nodule de petite taille, le plus souvent de manière unilatérale, bien que des cas d’adénomes affectant les deux surrénales ont été décrits.
(3)
Les adénomes non sécrétants
Les adénomes peu ou non sécrétants sont des tumeurs très fréquentes de la surrénale, avec une prévalence estimée à 5% de la population générale. Ces tumeurs sont généralement découvertes fortuitement lors d’examens d’imagerie prescrits sans lien avec la surrénale - on parle d’incidentalomes (Barzon et al., 2003 ; Tabarin et al., 2008).
Entre les adénomes cortisoliques associés à un syndrome de Cushing franc et les adénomes sans activité endocrine, des tumeurs sécrétant peu de cortisol, associées à un syndrome de Cushing infraclinique - i.e. sans symptôme clinique - sont également trouvées fortuitement, complétant le spectre de la sécrétion du cortisol par les adénomes corticosurrénaliens.
c) Les hyperplasies et dysplasies de la
surrénale
Parfois, dans les cas des hyperplasies, c’est l’ensemble du cortex surrénalien qui est affecté, ce pour les deux surrénales. Ces hyperplasies peuvent être responsables d’une hypersécrétion ou être non sécrétantes. Différents sous-types sont connus.
(1)
Les hyperplasies dépendantes de l’ACTH circulant
(a)
La maladie de Cushing et les sources
ectopiques d’ACTH
La maladie de Cushing est la principale cause de syndrome de Cushing. Elle compte en effet pour plus de 70% des causes de cet hypercorticisme (Newell-Price et al., 2006). Elle est due à un excès d’hormone corticotrope du fait d’une sécrétion accrue d’ACTH par un adénome hypophysaire bénin, dit corticotrope. Plus rarement, des tumeurs endocrines développées en dehors de l’hypophyse sécrètent un excès d’ACTH. On parle alors de source ectopique d’ACTH. Le développement accru du cortex surrénalien et l’excès de production de cortisol résulte de cette hypersécrétion d’hormone corticotrope.
Si la maladie de Cushing n’est - d’un point de vue corticosurrénalien - pas une tumeur à proprement parler, il s’agit néanmoins d’une hyperplasie du fait d’une prolifération anormale et accrue des cellules du cortex surrénalien. C’est pourquoi je parlerai plus globalement de « lésions » bénignes plutôt que de tumeurs bénignes lorsque je me réfèrerai également aux maladies de Cushing.
(b) L’hyperplasie congénitale des surrénales
Les hyperplasies congénitales des surrénales sont dues à des défauts de tout ou partie de la synthèse des hormones stéroïdes (El-Maouche et al., 2017). La majeure partie des cas est due à des défauts des enzymes stéroïdiennes du cortex surrénalien, à savoir des mutations de CYP21A2, de HSD3B2, de CYP11B1 ou plus rarement deCYP17A1.
En absence de sécrétion de cortisol, le complexe hypothalamo-hypophysaire produit de manière accrue de l’ACTH par rétrocontrôle. Cela entraîne un développement accru du cortex surrénalien. Outre les déficits en glucocorticoïdes, voire en
minéralocorticoïdes, on observe également un excès de production d’androgènes (sauf pour les déficiences en 17,20 lyase), du fait de l’accumulation des précurseurs en amont de la synthèse de cortisol (cf. figure sur la stéroïdogenèse corticosurrénalienne).
Des formes plus graves d’hyperplasies congénitales sont dus à des défaut de synthèse de l’ensemble des hormones stéroïdiennes, du fait de mutations inactivatrices de
STAR ou de CYP11A1 (Lin et al., 1995). On parle d’hyperplasies congénitales lipoïdes
des surrénales ; le développement des gonades est également impacté.
Je ne parlerai pas plus de ces lésions, qui n’ont pas fait l’objet de mon travail. Nous nous penchons en effet au laboratoire sur les tumeurs expliquant un excès d’hormones, et principalement le cortisol.
(2)
Les hyperplasies primitivement surrénaliennes
(a)
Les dysplasies micronodulaires
Les principales dysplasies micronodulaires sont les PPNAD (pour Primary
Pigmented Nodular Adrenal Dysplasia).
Ce sont des lésions rares affectant souvent des enfants et présentant des formes familiales. Elles sont associées à un hypercortisolisme sévère, parfois dans le cadre du complexe de Carney (Carney et al., 1985), caractérisé par d’autres affections comme des myxomes cardiaques, des nodules thyroïdiens, ou des lésions pigmentées de la peau.
Ces hyperplasies se présentent généralement sous la forme de petits nodules pigmentés, sur les deux cortex surrénaliens.
(b)
Les hyperplasies macronodulaires
primitives
Les hyperplasies macronodulaires primitives des surrénales sont caractérisées par un large épaississement des deux cortex surrénaliens, pouvant être accompagné de multiples macronodules. Elles sont de taille supra-centimétrique et ont été décrites pour la première fois dans les années 1960 (Kirschner et al., 1964),
Ces lésions sont rares, et comptent pour environ 1% des cas de syndrome de Cushing franc (Lacroix, 2009). Mais certaines formes, moins franches, peuvent être associées à un hypercorticisme moins marqué et à un syndrome de Cushing infraclinique.
Elles ont longtemps été nommées AIMAH, pour ACTH-Independant
Macronodular Adrenal Hyperplasia. Cependant, même si cet hypercorticisme est
indépendant de la stimulation par l’axe hypothalamo-hypophysaire ou d’une source ectopique d’ACTH, des études récentes ont montré une production intra surrénalienne d’hormone corticotrope, et donc la possibilité d’une stimulation de MC2R par effet paracrine/autocrine (Louiset et al., 2013). Ces lésions ont donc été renommées PMAH (pour Primary Macronodular Adrenal Hyperplasia).
B. Anomalies moléculaires des tumeurs
bénignes corticosurrénaliennes et apports de la
génomique
La génomique et les altérations moléculaires des tumeurs corticosurrénaliennes ont fait l’objet d’une revue que j’ai coécrite avec mon directeur de thèse ; cette revue figure en annexe de cette thèse.
Si des altérations de diverses voies de signalisation étaient déjà connues avant l’ère de la génomique - notamment les voies AMPc/PKA, Wnt/beta-caténine, et du calcium - le séquençage haut-débit ainsi que les puces SNP ont permis d’identifier de nouveaux acteurs de ces voies de signalisation dans la tumorigenèse, aussi bien bénigne que maligne.
Les méthodes « omics » modernes que sont le transcriptome, le méthylome ou le miRNome ont en outre amené de nouveaux éléments pour la connaissance de la physiopathologie de ces tumeurs, des outils de classifications moléculaires et des mécanismes alternatifs de tumorigenèse et de régulation de la sécrétion hormonale.
1. Voies de signalisation dérégulées dans les
tumeurs surrénaliennes et apports du
séquençage haut-débit
a) L’activation de la voie AMPc/PKA associée
aux tumeurs sécrétant du cortisol
(1)
Connaissance de l’activation de la voie dans le
syndrome de Cushing avant la génomique
(a)
Activation de la PKA par l’hormone
corticotrope (ACTH) et MC2R
L’hormone corticotrope ou ACTH est produite par l’hypophyse à partir du précurseur pro-opiomelanocortin (codé par POMC), sous le contrôle de la CRH (Cortisol Releasing Hormone) produite par l’hypothalamus (Feek et al., 1983). L’ACTH est le ligand du récepteur Melanocortin 2 receptor (MC2R), dont la découverte date des années 1930 (Collip et al., 1933).
L’action de l’ACTH via MC2R et son partenaire MRAP induit une prolifération accrue au sein du cortex et la différenciation en cellules de la zF (Hornsby, 1984). Les hyperplasies surrénaliennes dépendantes de l’ACTH circulant, dans le cadre de maladie de Cushing ou des hyperplasies congénitales des surrénales, sont la preuve du rôle de l’ACTH dans la prolifération corticosurrénalienne. Des mutations inactivatrices de MC2R et plus récemment de MRAP, sont liés à des cas de familial
glucocorticoid deficiency (FGD) (Génin et al., 2002 ; Metherell et al., 2005); causant
une forte résistance à l’ACTH, une absence de production de cortisol par les surrénales et un défaut de développement corticosurrénalien. MRAP est une protéine accessoire, nécessaire aux processus de modifications post-traductionnels de MC2R et à son mouvement vers la membrane plasmatique (Sebag and Hinkle, 2007).
La fixation de l’ACTH sur MC2R active la voie de l’AMPc, et induit l’expression de gènes permettant la sécrétion de cortisol : gènes du transport du cholestérol, de synthèse du cholestérol, de STAR, ou des enzymes de la stéroïdogenèse (Ganong et al., 1974)., Le cortisol lui-même exerce un rétrocontrôle négatif sur l’hypothalamus et sur la CRH (Feek et al., 1983).