© Maxime Gagnon, 2019
Stratégie analytique pour la résolution de
problématiques reliées à la présence de fondant dans
une solution préparée par fusion
Mémoire
Maxime Gagnon
Maîtrise en chimie - avec mémoire
Maître ès sciences (M. Sc.)
Stratégie analytique pour la résolution de problématiques reliées à la présence de fondant dans une solution préparée par fusion
Mémoire
Maxime Gagnon
Sous la direction de :
iii
Résumé
L’analyse par plasma à induction par micro-ondes (MIP) est l’une des techniques les plus prometteuses dû à ses nombreux avantages et à sa simplicité d’utilisation pour la détermination de métaux, incluant ceux pertinents pour le domaine minier. Cependant, l’introduction d’échantillons solide nécessite une mise en solution complète ou partielle.
De nombreuses études comparatives ont été publiées concernant ces modes de mises en solution pour divers analytes. Pour des matrices et des éléments réfractaires, la fusion alcaline permet généralement une mise en solution complète et de façon rapide. Néanmoins, la solubilisation par fusion possède certaines limitations en lien avec la mesure des métaux. La principale étant qu’une fois l’échantillon dissout par fusion dans l’acide, la charge en sel provenant du fondant est très élevée dans la solution obtenue, ce qui affecte grandement les performances d’ionisation du plasma. Considérant l’émergence d’instrumentation de type MIP dans les domaines traditionnels d’applications de la fusion, la présence d’espèces facilement ionisables (EFI) dans un plasma à plus faible température peut résulter en un biais analytique important.
Par conséquent, l’objectif de ce mémoire est de réduire la charge en sel des échantillons dissout par fusion avant leur introduction dans le système d’analyse afin de minimiser les coûts de la main-d’œuvre et le temps nécessaire à l’entretien de l’instrumentation tout en bonifiant les performances analytiques. Ceci sera effectué à l’aide d’approches analytiques basées sur des principes de séparation chromatographiques. Initialement, les profils d’élutions de divers éléments d’intérêts seront étudiés et analysé par ICP-AES. Par la suite, les paramètres des méthodes développées seront optimisés et validée à l’aide d’un matériel de référence certifié (MRC). Finalement, plusieurs paramètres analytiques, dont les limites de détection (LDM) et les limites de quantification des méthodes développées, seront déterminés par MIP-AES.
iv
Summary
Microwave induction plasma (MIP) is one of the most promising techniques because of its many advantages and its ease of use for determining metals, including those relevant to the mining industry. However, the introduction of solid samples requires complete or partial dissolution. Numerous comparative studies have been published concerning these dissolution modes for various analytes. For matrices and refractory elements, the alkaline fusion generally allows a fast and complete dissolution. Nevertheless, fusion solubilization has some limitations in connection with the measurement of metals. The main one being that once the sample melts in the acid, the salt load from the flux is very high in the resulting solution, which greatly affects the ionization performance of the plasma. Considering the emergence of MIP type instrumentation in traditional areas of fusion applications, the presence of easily ionizable species (EIS) in a lower temperature plasma can result in an important analytical bias. Therefore, the purpose of this study is to reduce the salt load of melt-dissolved samples prior to introduction into the analysis system to minimize labor costs and the time required for maintenance instrumentation while improving analytical performance. This will be done using analytical approaches based on chromatographic separation principles. Initially, the elution profiles of various elements of interest will be studied and analyzed by ICP-AES. Subsequently, the parameters of the methods developed will be optimized and validated using a certified reference material (CRM). Finally, several analytical parameters, including the detection limits (DL) and the quantification limits (QL) of the methods developed, will be determined by MIP-AES.
v
Table des matières
Résumé ... iii
Summary... iv
Table des matières ... v
Liste des tableaux ... vii
Liste des figures ... viii
Abréviations ... x
Remerciements ... xii
Introduction ... 1
Objectif du mémoire ... 6
Chapitre 1 – Théories et principes ... 7
1.0 Types de mise en solution ... 7
1.1 L’essai pyrognostique ... 8
1.2 Fusion alcaline ... 10
1.2.1 Tétraborate de lithium et métaborate de lithium ... 10
1.2.2 Peroxyde de sodium ... 11
1.2.3 Système de fusion automatisé ... 13
1.3 Appareils utilisés ... 14
1.3.1 Spectroscopie d’émission atomique au plasma à couplage inductif (ICP-AES) 14 1.3.2 Spectroscopie d’émission atomique à plasma micro-ondes (MP-AES) ... 17
1.3.3 Spectrométrie d’émission atomique à plasma à couplage inductif coupler à un spectromètre de masse en tandem (ICP-MSMS) ... 20
1.4 Inconvénients de la fusion ... 23
1.5 Techniques de séparation ... 24
1.5.1 Extraction liquide-liquide (ELL) et extraction liquide-liquide sur phase solide 25 1.5.2 Chromatographie d’échange ionique (EI) ... 26
1.5.3 Extraction sur phase solide (SPE) ... 28
1.6 Stratégie utilisée ... 30
1.6.1 Pour les platinoïdes ... 30
1.6.2 Ligand : Thiourée ... 33
1.6.3 Métaux communs ... 34
Chapitre 2 – Méthodologie ... 36
2.0 Réactifs et conditions instrumentales ... 36
2.1 Réactifs ... 36
2.2 Mise en solution des échantillons ... 36
vi
2.4 Matériel de référence certifié (MRC) OREAS-683 ... 42
2.5 ICP-AES ... 44
2.6 MP-AES ... 45
2.7 ICP-MS/MS ... 45
Chapitre 3 – Résultats et discutions ... 47
3. Développement d’une technique de séparation des platinoïdes en présence de peroxyde de sodium ... 47
3.1 Impact de la charge en sel sur la sensibilité instrumentale de l’ICP-AES et du MP-AES ... 47
3.2 Séparation des platinoïdes et du sodium par chromatographie ioniques ... 49
3.2.1 Impact de la quantité de fondant utilisé lors de la fusion sur les rendements de séparation ... 53
3.2.2 Impact de la teneur en HCl sur la rétention ... 55
3.2.3 Profils d’élution de l’or et du palladium par chromatographie anionique .... 58
3.2.4 Comparaison des courbes de Pt en fonction des facteurs de dilution par ICP-AES et MP-ICP-AES ... 60
3.3 Développement d’une technique de séparation des métaux communs en présence de peroxyde de sodium ... 61
3.3.1 Choix du tampon et du pH... 61
3.3.2 Analyse du matériel de références certifiés OREAS-683 ... 65
3.4 Tentative de couplage des deux méthodes ... 66
3.4.1 Modification du tampon – Saturation de la solution d’acétate d’ammonium en chlorure ... 71
3.4.2 Modification du tampon – Ajustement du pH des échantillons sans acétate d’ammonium ... 74
3.5 Étude de la capacité de la résine Dowex 1x8 et Nobias PA-1 ... 77
3.6 Analyse des métaux ciblés dans le fondant ... 80
3.7 Détermination des limites de détection et de quantification des 9 métaux d’intérêts par MP-AES ... 82
Conclusion ... 84
4.1 Retour sur les objectifs ... 84
4.2 Perspectives ... 85
vii
Liste des tableaux
Tableau 1 : Comparaison des caractéristiques de différents fondants. ... 12
Tableau 2 : Complexes formés par les platinoïdes en milieu aqueux et chloré. ... 31
Tableau 3 : Préparation des échantillons : Détails et produits nécessaires. ... 37
Tableau 4 : Paramètres d’utilisation du système de fusion Claisse LeNeo. ... 37
Tableau 5 : Méthode développée visant la séparation des platinoïdes d’une matrice chargée en sodium suite à une mise en solution par fusion au Na2O2. .... 39
Tableau 6 : Méthode développée visant la séparation des métaux communs d’une matrice chargée en sodium suite à une mise en solution par fusion au Na2O2. .... 41
Tableau 7 : Analytes et valeurs certifiées du MRC OREAS-683. ... 43
Tableau 8 : Conditions instrumentales pour l’analyse des platinoïdes et métaux communs du ICP-AES. ... 44
Tableau 9 : Conditions instrumentales pour l’analyse des platinoïdes et métaux communs du MP-AES... 45
Tableau 10 : Conditions instrumentales pour l’analyse des platinoïdes et métaux communs à l’état de trace dans le fondant par ICP-MS/MS. ... 46
Tableau 11 : Conditions étudiées lors du développement de la méthode Nobias PA-1. ... 62
Tableau 12 : Récupération obtenue à l’aide du MRC OREAS-683. ... 65
Tableau 13 : Méthode développée visant la séparation des platinoïdes et métaux communs d’une matrice chargée en sodium suite à une mise en solution par fusion au Na2O2. ... 67
Tableau 14 : Détermination de la capacité (Qe) (en mg/g) de la résine Dowex 1x8 et de la résine Nobias PA-1. ... 80
Tableau 15 : Concentrations en métaux traces analysées dans le peroxyde de sodium par ICP-MSMS. ... 81
Tableau 16 : Limite de détection (LDM) et limite de quantification (LQM) obtenues pour les méthodes Dowex 1x8 et Nobias PA-1 sur MP-AES. ... 83
viii
Liste des figures
Figure 1 : Valeur de la production minérale au Canada de 1999 à 2016 par
catégorie (en milliards de dollars canadiens) ... 1
Figure 2 : Nombre de publications par année déterminé à l’aide de Web of Science dont le sujet est « microwave plasma emission spectrometry ». ... 5
Figure 3 : Perle d’or obtenue une fois l’oxyde de plomb absorbé à même les parois du creuset de phosphate de calcium. ... 9
Figure 4 : Comparaison des pouvoirs oxydants de certains fondants versus leurs alcalinités. ... 13
Figure 5 : Schéma simplifié d’un polychromateur couplé à un spectromètre au plasma. ... 16
Figure 6 : Schéma simplifié du trajet parcouru par les ions dans un spectromètre de masse avec une cellule de collision-réaction. ... 22
Figure 7 : Schéma des différentes techniques de séparation ... 25
Figure 8 : Structure de la résine Dowex 1x8 ... 28
Figure 9 : Structure de la résine NOBIAS PA-1 et activation des groupements fonctionnels suivant une augmentation du pH. ... 30
Figure 10 : Diagramme de Pourbaix du platine ... 32
Figure 11 : Diagramme de Pourbaix du palladium ... 32
Figure 12 : Structure moléculaire du thiourée. ... 33
Figure 13 : Réduction et complexation d’un chlorocomplexe de platine (IV) en un complexe de platine (II) – thiourée. ... 34
Figure 14 : Schéma illustrant les étapes principales de la méthode de séparation des platinoïdes d’une matrice chargée en sodium. ... 40
Figure 15 : Schéma illustrant les étapes principales de la méthode de séparation des métaux communs d’une matrice chargée en sodium. ... 42
Figure 16: Analyse du Platine par ICP-AES et MP-AES en fonction de la nature de la matrice. ... 47
Figure 17: Histogramme d’élution du platine et du sodium selon leurs portions d’élution à l’aide d’une solution de thiourée 0,2M. (C : Chargement, L : Lavage). 50 Figure 18: Analyse du Platine par MP-AES en fonction de la nature de la matrice. ... 51
Figure 19: Histogramme d’élution du platine et du sodium selon leurs portions d’élution avec une solution de thiourée à une température de 25°C et 60°C (C : Chargement, L : Lavage). ... 52
Figure 20: Histogramme d’élution du platine et du sodium selon leurs portions d’élution dépendamment la quantité de fondant utilisé lors de la fusion à une température de 25°C (C : Chargement, L : Lavage). ... 54
Figure 21: Histogramme d’élution du cuivre selon la concentration en chlorures à une température de 25°C (C : Chargement, L : Lavage). ... 56
Figure 22: Histogramme d’élution des métaux communs selon les portions d’élution avec une solution de thiourée à une température de 25°C (C : Chargement, L : Lavage). ... 57
ix
Figure 23: Histogramme d’élution du Fer selon la concentration en chlorures à une température de 25°C (C : Chargement, L : Lavage). ... 58 Figure 24: Histogramme d’élution de l’or, du platine, du palladium et du sodium selon leurs portions d’élution avec une solution de thiourée à une température de 25°C (C : Chargement, L : Lavage). ... 59 Figure 25: Comparaison de la sensibilité d’une courbe de platine selon la dilution de la matrice par ICP-OES. ... 60 Figure 26: Analyse de la sensibilité d’une courbe de platine selon la dilution de la matrice par MP-AES. ... 61 Figure 27: Histogramme d’élution du nickel selon le type d’éluant à différents pH (C : Chargement, L : Lavage). ... 62 Figure 28: Histogramme d’élution des métaux communs et du sodium selon leurs portions d’élution avec une solution de HCl 1M (C : Chargement, L : Lavage). ... 64 Figure 29 : Schéma illustrant les étapes principales des méthodes couplées visant la séparation des métaux communs et platinoïdes d’une matrice chargée en
sodium. ... 68 Figure 30 : Histogramme d’élution des métaux communs suite au couplage des méthodes Dowex 1x8 et Nobias PA-1 ... 69 Figure 31 : Histogramme d’élution des platinoïdes suite au couplage des
méthodes Dowex 1x8 et Nobias PA-1 ... 70 Figure 32 : Histogramme d’élution des métaux communs suite au couplage des méthodes Dowex 1x8 et Nobias PA-1 en utilisant un tampon fortifié en chlorure. ... 72 Figure 33 : Histogramme d’élution des platinoïdes suite au couplage des
méthodes Dowex 1x8 et Nobias PA-1 en utilisant un tampon fortifié en chlorure. ... 73 Figure 34 : Histogramme d’élution des métaux communs suite au couplage des méthodes Dowex 1x8 et Nobias PA-1 en utilisant une solution fortifiée en
chlorure ajusté à pH 6. ... 75 Figure 35 : Histogramme d’élution des platinoïdes suite au couplage des
méthodes Dowex 1x8 et Nobias PA-1 en utilisant une solution fortifiée en
chlorure ajusté à pH 6. ... 76 Figure 36 : Graphique représentant le profil d’adsorption de la résine Dowex 1x8. ... 78 Figure 37 : Graphique représentant le profil d’adsorption de la résine Nobias PA-1. ... 78 Figure 38 : Graphique représentant le ratio de la masse de palladium sorbé sur la masse de résine Dowex (S)(mg/g) en fonction de la concentration à l’équilibre (mg/L). ... 79
x
Abréviations
(AAF) Spectroscopie d’absorption atomique flamme
(AAFG) Spectroscopie d’absorption atomique par four graphite (CO) Monoxyde de carbone
(DVB) Divinyl-benzène
(ED3A) Acide ethylenediaminetriacétique (ELL) Extraction liquide-liquide
(ELLI) Extraction liquide-liquide ionique
(ELLS) Extraction liquide-liquide sur support solide (FD) Facteur de dilution
(H3BO3) Acide borique
(HC) Hydrocarbure
(HCl) Acide chlorhydrique
(ICP) Spectrométrie d’émission atomique à couplage inductif (ICP-MS) Spectrométrie de masse
(ICP-OES) Spectroscopie optique d’émission (IDA) Acide iminodiacétique
(LDM) Limite de détection de la méthode (LiBO2) Métaborate de lithium
(Li2B4O7) Tétraborate de lithium
(LQM) Limite de quantification de la méthode
(MP-AES) Spectromètre d’émission atomique à plasma micro-onde (MRC) Matériel de référence certifié
(Na2B4O7) Borax
(NaCO3) Carbonate de sodium
(Na2O2) Peroxyde de sodium
(NOx) Oxydes d’azote
(PbO) Oxyde de plomb (II) (PS) Polystyrène
(SPE) Extraction sur phase solide
xi
Success is not final, failure is not fatal: it is the courage to continue that counts. Sir Winston Churchill
xii
Remerciements
J’aimerais remercier le professeur Dominic Larivière de m’avoir accueilli dans son laboratoire ainsi que toute l’équipe ; Alexa, Guillaume, Claire, Julie, Anthony, Samantha, Elisabeth et Audrey. Je souhaite aussi remercier Keven Turgeon et M. Serge Groleau pour leurs expertises concernant l’instrumentation. De plus, je veux aussi remercier MITACS et Claisse, et plus particulièrement Mathieu Bouchard, Mathieu Hamel, Janice Pitre et Mélanie Bédard pour leurs soutiens dans le cadre de ce projet.
Finalement, je ne peux pas assez remercier celle qui me supporte et m’appuie depuis toutes ces années, merci à toi, Véro, merci pour tout. Sans toi, je ne sais vraiment pas comment j’aurais pu faire tout ça. Merci de ta présence, que ce soit lors de nos réussites ou encore dans des moments plus difficiles, je suis si heureux de t’avoir à mes côtés. Je t’aime plus que tout !
1
Introduction
L’industrie minière est l’un des secteurs économiques d’importance au Canada. Le Canada est un leader mondial dans la production minière, autant du point de vue financier que par son expertise [1]. En effet, comme l’indique la figure 1, la production minière a rapporté au Canada plus de 40 milliards de dollars en 2016 dont plus de la moitié provenaient des métaux.
Figure 1 : Valeur de la production minérale au Canada de 1999 à 2016 par catégorie (en milliards de dollars canadiens) [2]
Les métaux comme l’or, l’argent, le platine, le fer, le cuivre ou encore le nickel sont des métaux essentiels à la société industrielle moderne [1]. Ils permettent la fabrication d’une multitude d’appareils couramment utilisés, allant des pièces automobiles aux composantes de nos appareils électroniques.
2
Deux familles de métaux sont d’intérêts pour les minières canadiennes, soit les platinoïdes et les métaux communs. La première famille inclut le ruthénium (Ru), le rhodium (Rh), le palladium (Pd), l’osmium (Os), l’iridium (Ir) et platine (Pt)[3]. Ces métaux possèdent des propriétés physiques et chimiques bien uniques qui les rendent économiquement intéressants. En effet, comme ils sont chimiquement inertes, malléables, ainsi que très stable, ils sont couramment utilisés dans les domaines biomédicaux et la joaillerie [4]. Ils sont également très utiles comme catalyseur pour certaines applications[3–6] ou encore dans des médicaments contre le cancer [7]. Ils sont néanmoins peu abondants dans la croute terrestre [8-9], ce qui fait augmenter leur valeur marchande.
À titre d’exemple, les voitures doivent être munies de convecteur catalytique afin de réduire les émissions en monoxyde de carbone (CO), en oxydes d’azote (NOx) et en hydrocarbures (HC). L’installation de convecteur
catalytique a commencé en 1973 au Japon, en 1975 aux États-Unis et en 1986 en Europe[9]. Les métaux contenus dans ces dispositifs sont principalement le platine ainsi que le palladium et le rhodium. Ces convecteurs catalytiques possèdent une période effective entre 40 000 et 80 000 km dépendamment de leur qualité. Ils deviennent de moins en moins efficaces suite à la grande variation de température que subit le convecteur catalytique. La surface active des catalyseurs à l’intérieur du convecteur en est diminuée et, par conséquent, ils doivent être remplacés. On estime à plusieurs millions le nombre de convecteurs changés par an, chacun contenant de 0,5 à 1 g de platine et de 1 à 2 g de platinoïdes[10].
D’autres métaux, extraits à partir de minerais plus riches, revêtent également un intérêt commercial pour l’industrie minière, essentiellement en lien avec le tonnage annuel produit. Ce sont des métaux tels que le magnésium (Mg), le chrome (Cr), le cobalt (Co), le cuivre (Cu), le nickel (Ni) et le fer (Fe). Ces éléments disposent de plusieurs propriétés qui en font des éléments recherchés dans l’industrie minière. Ils sont par exemple utilisés
3
dans des alliages pour rendre un matériau solide et léger, comme dans de cas du magnésium, ou encore lui donner une résistance accrue à la corrosion comme dans le cas du chrome. Le cobalt est utilisé dans le domaine des aimants de type AlNiCo ou encore incorporé dans un alliage afin d’augmenter sa résistance thermique. Nul besoin de mentionner tous les domaines d’application dans lesquels le fer est utilisé pour comprendre sa pertinence dans notre environnement urbain. De faible coût et abondant, il est utilisé dans divers domaines, de la production de l’acier aux catalyseurs en chimie organique. Finalement, dans le cas du nickel, il est utilisé comme agent anticorrosion lorsqu’incorporé dans un alliage ou encore dans les batteries.
Dans le cadre de ce projet de maîtrise, ces métaux seront référés comme appartenant au groupe des métaux communs.
Comme la valeur réelle des gisements miniers est basée sur l’évaluation de la teneur des métaux qu’ils contiennent, la quantification adéquate des analytes devient donc à la fois un enjeu analytique et économique. L’exemple de la compagnie Bre-X[11] qui a fourni des données altérées relatives aux teneurs en or dans leurs gisements est un rappel de l’importance d’une métrologie adéquate dans le domaine de la prospection minière.
En ce sens, les développements analytiques permettant d’assurer un haut degré de justesse et de précision lors des analyses élémentaires minéralogiques sont un enjeu critique pour l’industrie minière. Ceci s’applique à la fois à la préparation des échantillons qu’à la mesure elle-même.
Bien que souvent sous-estimée dans le processus analytique, la mise en solution des échantillons, souvent nécessaire lors de l’utilisation des techniques d’analyse élémentaire communes, demeure un paramètre clé dans un processus de métrologie approprié. À titre d’exemple, les platinoïdes ont tendance à former des oxydes réfractaires qui sont peu
4
solubles dans les conditions de traitement d’échantillons usuelles et requièrent des stratégies plus efficaces telles que la fusion alcaline[12].
Il existe présentement une multitude de techniques d’analyse élémentaire utilisées dans tout autant de matrices variées [13]. L’une des techniques d’analyse les plus communes utilisée dans le domaine minier est l’analyse par spectrométrie de fluorescence des rayons X (XRF). Cette technique est rapide, relativement simple et nécessite que peu de préparation d’échantillon puisqu’il est possible d’analyser l’échantillon à l’état solide. La précision et la justesse de cette méthode dépendent énormément de l’homogénéité de l’échantillon [14] en comparaison à des techniques telles que l’absorption atomique à flamme (AAF) ou encore par spectrométrie d’émission atomique à couplage inductif (ICP) [13]. Ce faible degré de précision peut avoir de grandes répercussions financières surtout dans les cas où il demeure très rentable d’exploiter un gisement minier à des teneurs marginales comme c’est le cas avec les platinoïdes. Une technique alternative qui s’est implantée au cours des trente dernières années dans le domaine de l’analyse minérale est l’utilisation d’instrument ICP. À titre d’exemple, Zawisza et coll.[15] ont montré que plus de 60% des analyses d’éléments de terre rare pour le domaine minier sont maintenant faites par spectroscopie optique d’émission (ICP-OES) ou spectrométrie de masse (ICP-MS). L’utilisation de plasma comme source d’atomisation, d’excitation et d’ionisation est l’objet d’étude de plusieurs groupes de recherche et ce, depuis nombre d’années [16-18]. Une nouvelle technique d’émission tend néanmoins à vouloir émerger depuis quelques années, soit l’émission atomique à plasma micro-onde (MP-AES). Bien que l’utilisation d’un plasma induit par micro-onde usant d’une fréquence de 2,45 GHz remonte à 1951 [19], cette technique est beaucoup plus explorée depuis l’arrivé sur le marché du MP-AES 4100 d’Agilent technologies en 2011. En
5
effet, il est possible de remarquer que le nombre de publications à ce sujet est en augmentation depuis les deux dernières décennies.
Figure 2 : Nombre de publications par année déterminé à l’aide de Web of Science dont le sujet est « microwave plasma emission spectrometry ».
Comme la technique d’ICP, le MP-AES nécessite la mise en solution complète des échantillons afin d’avoir une quantification juste et précise. Pour ce faire, la technique de mise en solution privilégiée est la mise en solution par fusion au peroxyde de sodium (Na2O2). Cette technique est
rapide et très efficace lors de la digestion d’échantillons géologiques [20-23]. Il existe cependant quelques défis à relever entre le couplage des techniques de mise en solution par fusion et les techniques de mesure par spectroscopie d’émission atomique par plasma micro-onde.
N o m b re d e p u b li ca ti o n s Années
6 Objectif du mémoire
Ce mémoire de maîtrise porte donc sur l’élaboration d’une stratégie analytique pour la résolution de problématiques reliées au couplage entre la fusion alcaline et la MP-AES. Plus spécifiquement, les problèmes liés à la quantité et qualité des sels dissouts présents dans un échantillon analysé par spectroscopie d’émission atomique à plasma micro-ondes après une mise en solution par fusion au peroxyde de sodium (Na2O2). L’utilisation de
diverses résines échangeuses d’ions est au cœur même de plusieurs groupes de recherche[24]–[26] œuvrant dans le développement de méthodes d’analyses visant la ségrégation d’une ou de plusieurs espèces chimiques. Ce projet de maîtrise a donc comme objectif la séparation du sodium (Na) ayant pour origine le fondant utilisé lors de la fusion au Na2O2 en utilisant
des résines échangeuses d’ions. En premier lieu, l’utilisation de la résine anionique Dowex 1X8 (Millipore Sigma, USA) aura comme éléments visés le groupe des platinoïdes, soit l’or (Au), le platine (Pt) et le palladium (Pd). En second lieu, la résine Nobias PA-1 (Hitachi High-Technologies, Japon) sera utilisée afin de séparer le sodium du groupe des métaux communs, comprenant le chrome (Cr), le cobalt (Co), le cuivre (Cu), le fer (Fe), le magnésium (Mg), et finalement le nickel (Ni).
7
Chapitre 1 – Théories et principes
Lors d’analyses élémentaires, la préparation d’échantillon est certainement l’une des étapes les plus importantes. En effet, que ce soit pour une analyse par spectroscopie ou spectrométrie, la mise en solution complète de l’échantillon est souvent nécessaire et primordiale afin d’obtenir des résultats valides et précis. Il existe plusieurs méthodes de mise en solution couramment utilisées afin d’atteindre cet objectif. Ces méthodes seront discutées dans ce chapitre. Par la suite, il sera question de diverses techniques d’analyse utilisées dans le domaine de l’analyse d’échantillons géologiques et des contraintes reliées à ce type d’analyse. Il sera, entre autre, sujet de la problématique liée au couplage du type de mise en solution utilisé avec les techniques analytiques visées. Finalement, il sera ensuite question des pistes de solution et des stratégies planifiées afin de remédier aux problématiques et d’atteindre les objectifs du projet.
1.0 Types de mise en solution
Pour commencer, que ce soit pour une analyse par spectroscopie absorption atomique flamme (AAF), par four graphique (AAFG) ou encore par spectrométrie d’émission atomique à plasma (ICP-OES), il est souvent requis d’effectuer une mise en solution afin d’obtenir une meilleure homogénéité de l’échantillon et de s’assurer de la compatibilité avec le système d’introduction de l’instrument. Lorsqu’il est question d’une digestion d’échantillon fortement minéralisé, il est impératif que l’échantillon solide soit complètement dissout. En effet, le solide doit être totalement dissout afin de diminuer les risques de sous-évaluation de la quantité réelle des éléments analysés. De plus, la nature chimique de l’échantillon aura une grande influence sur la technique de mise en solution préconisée. En effet, certaines composantes des échantillons seront plus difficiles à digérer dû à leurs caractéristiques propres telles que leur
8
caractère réfractaire ou leur capacité à former des composés volatils. Ces caractéristiques sont particulièrement présentes dans les échantillons miniers ayant été soumis, sur une longue échelle de temps, à des processus de minéralisation et de dissolution. Il existe un grand nombre de stratégies de mise en solution en utilisation ou en développement dans le secteur minier, mais deux se démarquent, soit l’essai pyrognostique et la fusion alcaline.
1.1 L’essai pyrognostique
L’une des plus anciennes méthodes de séparation de métaux précieux est certainement l’essai pyrognostique (fire assay). En fait, cette technique remonte à aussi loin que l’Antiquité et avait pour objectif de purifier l’or et l’argent. Il est possible d’utiliser cette technique de purification préliminaire pour la mise en solution d’éléments tels que les platinoïdes.
Les concepts de bases des essais pyrognostiques sont relativement simple[3]. Pour commencer, l’échantillon minéral est mélangé avec un fondant, principalement du carbonate de sodium (NaCO3), du borax
(Na2B4O7), de la silice (SiO2) ou encore du peroxyde de sodium (Na2O2). Une
quantité d’oxyde de plomb (PbO) est ajoutée au mélange et joue le rôle de collecteur. Les métaux désirés seront solubles dans le plomb métallique fondu alors que le reste de la matrice se retrouvera dans la scorie (slag). Finalement, le mélange est chauffé à 1100°C afin de mettre le mélange sous sa forme liquide et de favoriser le transfert des éléments. L’essai pyrognostique se base sur la différence de gravité spécifique des deux phases liquides formées lors de l’étape de liquéfaction, soit celle de la scorie et celle de l’alliage de plomb métallique. Une fois la scorie et l’alliage de plomb séparés, ce dernier est solubilisé à l’aide d’une fusion oxydant rigoureusement contrôlée dans une coupelle en phosphate de calcium. L’oxyde de plomb (II) (PbO) est alors absorbé dans les parois de la coupelle
9
et les métaux précieux forment, quant à eux, de petites billes au fond du creuset. Il est possible, par la suite, de dissoudre ces billes dans de l’acide nitrique afin de les analyser par voie humide.
Figure 3 : Perle d’or obtenue une fois l’oxyde de plomb absorbé à même les parois du creuset de phosphate de calcium. [27]
Le principal avantage de cette méthode de séparation et mise en solution est la possibilité d’utiliser une masse d’échantillons relativement élevée (10-50 grammes)[12] et de la convertir en une petite bille de métaux précieux. De plus, elle ne nécessite pas de matériel spécifique ou dispendieux.
Cependant, les essais pyrognostique nécessitent un personnel hautement qualifié[3,20] afin de mélanger adéquatement les divers fondants ou d’obtenir les conditions et températures de fusion optimales. L’introduction d’une grande quantité de sels provenant du fondant peut également mener à une contamination involontaire de l’échantillon en plus d’augmenter l’intensité des blancs lors de l’analyse. Un problème majeur avec cette technique est que la récupération des métaux nobles (platinoïdes, or et argent) par l’oxyde de plomb est fortement dépendante de la solubilité de ces derniers [3,12]. En effet, il est possible d’obtenir d’excellents rendements pour l’or, l’argent, le platine et le palladium alors que ceux pour le ruthénium, rhodium, et iridium sont plus problématiques. L’osmium a d’ailleurs tendance à former un composé volatil (OsO4)[28] lorsqu’il est
10 1.2 Fusion alcaline
La mise en solution par fusion alcaline est certainement l’une des méthodes les plus agressives permettant la dissolution complète et rapide d’un échantillon solide. Comme pour la partie initiale des essais pyrognostique, l’échantillon est mélangé à un fondant tel que le peroxyde de sodium (Na2O2), le carbonate de calcium (Na2CO3) ou encore un mélange de
tétraborate de lithium (Li2B4O7) et de métaborate de lithium (LiBO2). Durant
la fusion, l’analyte d’intérêt vient se lier à l’anion du fondant. Bien que cette réaction ne soit pas favorable, le grand excès de fondant en comparaison avec la quantité d’échantillon et la haute température du milieu de fusion facilite une telle réaction. La suite de la réaction dépend grandement du fondant lui-même.
1.2.1 Tétraborate de lithium et métaborate de lithium
Dans le cas du métaborate de lithium (LiBO2), chauffé à 1100°C dans un
creuset de platine, l’anion borate vient se lier à l’ion métallique, formant ainsi une perle de fusion. Il est possible de laisser refroidir cette perle afin de former un disque pouvant servir à l’analyse des métaux présents dans cette même perle par technique de spectrométrie de fluorescence des rayons X (XRF). Cette technique de préparation possède plusieurs avantages[29]. En effet, il est possible d’éliminer complètement les différences dues à l’effet minéralogique entre les échantillons étant donné que les perles de fusion sont totalement homogènes. Cette homogénéité améliore grandement la reproductibilité et la précision des méthodes. Il est aussi possible de dissoudre le mélange de fusion dans une solution d’acide diluée. En effet, le produit formé du métal lié au borate n’est pas soluble dans l’eau, mais une fois en contact avec un acide dilué, la formation d’acide borique (H3BO3)
viendra favoriser la réaction de dissolution d’une perle comme démontré avec l’équation suivante :
11
( ) ( )+ 6 ( )+ 4 ( ) → ( )+ 4 ( )+ 2
Le métaborate de lithium et le tétraborate de lithium sont très polyvalents. En effet, peu de matrices résistent à leurs pouvoirs de digestion mise à part les sulfures et certains alliages métalliques. Cette capacité vient du fait qu’ils ne possèdent pas le même caractère acido-basique. Le tétraborate a un caractère plus acide alors que le métaborate possède, pour sa part, un caractère plus alcalin. Donc, dépendamment du type de matrice, il est possible de choisir la bonne espèce ou encore de faire un mélange à différent ratio des deux fondants afin d’obtenir des conditions de dissolution adéquates.
1.2.2 Peroxyde de sodium
Le peroxyde de sodium (Na2O2) est certainement l’un des fondants les
plus agressifs pour une matrice. Étant un oxydant, il a la capacité de dissoudre un grand nombre de matrices, des matériaux réfractaires aux éléments purs en passant par les sulfures. Il y a quelques années, Chand et coll. [30] ont d’ailleurs comparé l’efficacité de digestion entre la fusion alcaline et la digestion assistée par microonde et ils ont conclu que la première technique est plus efficace lors de la mise en solution d’élément réfractaire. Puisque le Na2O2 peut dissoudre un grand nombre de métaux,
incluant le Pt, l’utilisation de creusets faits de cet élément est à proscrire. Seuls les creusets de zirconium possèdent une résistance chimique modérée au Na2O2 et des traces de zirconium (Zr) sont détectables dans les solutions
finales après fusion. De plus, le Na2O2 a tendance à réagir violemment avec
les éléments facilement ionisables ou encore les matrices organiques. L’utilisation de température plus basse (600°C) et l’ajout de carbonate de calcium peuvent réduire ce type de réaction. Finalement, le mélange de fusion formé ne peut être utilisé pour l’analyse par XRF puisqu’il est non transparent et n’est pas stable à l’humidité de l’air. Il est cependant un
12
excellent candidat à la dissolution dans un acide dilué[22]. L’équation de la réaction de fusion est démontrée ci-dessous :
( )+ 2 ( ) → ( )+ ( )
Le composé de platine (IV) formé (Na2PtO3) est beaucoup plus soluble, ce qui
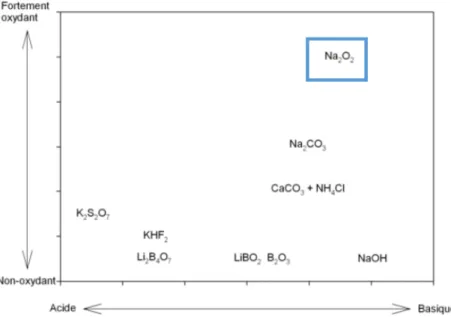
facilite grandement sa mise en solution dans une solution d’acide dilué. Le tableau 1 compare les caractéristiques de divers fondants tandis que la figure 4 compare le pouvoir oxydant des sels comparativement à leurs alcalinités. Le sel utilisé dans le cadre de ce projet est le peroxyde de sodium (Na2O2) compte tenu de sa capacité à digérer les échantillons les plus
coriaces et de son fort pouvoir oxydant. Il existe plusieurs autres fondants disponibles commercialement, mais ils sont destinés à des usages très spécifiques par exemple le trioxyde de bore (B2O3) pour l’analyse de métaux
alcalins.
Tableau 1 : Comparaison des caractéristiques de différents fondants.
Fondant Types de creuset Température de fusion (°C) Type d’échantillon dissout Na2CO3 Pt 850 Silicates LiBO2 Pt 1000-1100 Tout sauf sulfure et métaux Li2B4O7 Pt 1000-1100 Tout sauf sulfure et métaux Na2O2 Zr, Ni ou Fe 600 Tout
13
Figure 4 : Comparaison des pouvoirs oxydants de certains fondants versus leurs alcalinités. [31]
L’un des avantages de l’utilisation de la technique de mise en solution par fusion est certainement le temps de digestion. En effet, la fusion ne requiert que quelques minutes. De plus, il n’est plus nécessaire d’utiliser des acides nocifs tels que l’acide fluorhydrique (HF) puisque la dissolution du produit après fusion peut se faire dans une solution aqueuse d’acide dilué. Galindo et coll[23] ont démontré que la fusion au peroxyde offre les mêmes bénéfices en termes d’efficacité que la dissolution à l’aide du HF, mais sans les inconvénients associés à ce dernier.
1.2.3 Système de fusion automatisé
Un des défis historiques de la dissolution par fusion était l’utilisation de produits chimiques en présence de flammes et d’importante source de chaleur. Ce mélange de conditions d’exploitation augmentait le risque d’accident pour l’utilisateur. Au début des années 80, l’apparition
14
d’instruments automatisés et l’utilisation, depuis plus d’une décennie, de fours électriques en remplacement des brûleurs au propane ont fait de la fusion alcaline automatisée une méthode sécuritaire, reproductible et rapide. À l’aide d’un système de fusion automatisé, il est possible de préparer des échantillons par fusion pour des analyses XRF, AAF ou encore par ICP. Il est donc possible de préparer une large gamme d’échantillons, des échantillons miniers aux échantillons pharmaceutiques en passant par les catalyseurs et les alliages.
1.3 Appareils utilisés
Tel que démontré par Zawisza et coll.[15], une proportion importante des analyses dans le domaine minier s’effectue maintenant par spectrométrie à plasma et cette tendance semble s’accélérer. Dans les prochains paragraphes, nous décrierons les trois principaux appareils les plus couramment utilisés soit le spectromètre d’émission atomique à plasma à couplage inductif (ICP-OES), le spectromètre de masse à plasma à couplage inductif (ICP-MS) et, bien entendu, le spectromètre d’émission atomique à plasma micro-onde (MP-AES).
1.3.1 Spectroscopie d’émission atomique au plasma à couplage inductif (ICP-AES)
Les premières applications analytiques de la spectroscopie atomique remontent en 1776 avec les travaux de Volta. Par la suite, Kirchoff et Bunsen (1859) développèrent la technique afin d’exciter les métaux à l’aide d’une flamme. Par contre, seulement une petite quantité des atomes de métaux et métalloïdes se retrouvent sous une forme excitée aux températures typiques des flammes, soit 3000 °K [32]. Dans les années 1960, il est envisagé d’utiliser les plasmas comme milieu d’atomisation et c’est après seulement dix ans plus tard que les premiers appareils commerciaux apparaissent sur le marché. Ils sont cependant très dispendieux. Il faudra attendre au début des années 1990 pour observer leur véritable expansion comme appareils
15
de routine avec l’arrivée des détecteurs à semi-conducteurs bidimensionnels, qui exploitent le potentiel multiélémentaire et simultané de la technique. Aujourd’hui, la spectroscopie d’émission atomique est certainement l’une des techniques les plus répandues pour les analyses quantitatives de métaux et métalloïdes dans les laboratoires.
Afin de produire une analyse, l’échantillon doit absolument être sous forme aqueuse, sauf pour les appareils munis d’un système d’ablation laser. Ce type d’interface d’introduction d’échantillon possède l’avantage de diminuer considérablement le temps de préparation des échantillons puisqu’il n’est plus nécessaire de dissoudre ce dernier. Cependant, il est très difficile d’obtenir une quantification valable due à l’absence d’étalons appropriés et à l’hétérogénéité des échantillons solides[33-35]. Pour ces raisons, l’aspiration d’échantillons dissouts demeure l’approche favorisée pour l’analyse de matrices par spectroscopie atomique. L’échantillon aqueux est dirigé vers le nébuliseur qui crée de fines gouttelettes, lesquelles sont produites dans une chambre de nébulisation et finalement introduite dans le plasma à l’aide d’un débit d’argon.
Le plasma est produit par les collisions entre les électrons et le gaz constituant le plasma, c’est-à-dire l’argon ionisé (Ar+). Ces ions circulent
dans un champ électrique produit par une bobine Tesla génératrice de radiofréquence à 25-40 MHz. C’est la « friction » des électrons et cations Ar+
forcés de circuler dans ce champ magnétique, qui oscille à de très hautes fréquences, qui génère la chaleur intense du plasma (de l’ordre de 7 000 à 10 000 Kelvins). À cette température, les analytes peuvent se retrouver sous deux formes ; atomique ou ionique. Environ la moitié se retrouve sous forme atomique (raies de type « I ») alors que l’autre moitié se retrouve sous forme d’ion monovalent (raie de type « II »).
Une fois excités, les électrons des couches inférieures sont alors promus à un niveau électronique supérieur, mais seulement pour une courte période
16
de temps. En se relaxant, ces derniers émettent un photon ayant une longueur d’onde caractéristique pour chacun des analytes, basé sur les différentes transitions électroniques permises.
La séparation des longueurs d’onde est faite à l’aide de deux éléments dispersifs ; un réseau et un prisme. Il est donc possible de disperser les longueurs d’onde en deux dimensions. On obtient ainsi un système optique avec une grande résolution, particulièrement efficace dans la région des ultraviolets, où se retrouve la grande majorité des raies analytiques couramment utilisées en émission atomique.
Figure 5 : Schéma simplifié d’un polychromateur couplé à un spectromètre au plasma.
L’avantage principal de l’ICP-AES est certainement le fait qu’il soit multiélémentaire et permet la quantification sur un large domaine de concentration. La nature multiélémentaire du système de séparation et de détection augmente de beaucoup la cadence analytique de cet instrument. En effet, il ne nécessite que quelques minutes par échantillons, et ce, même si plusieurs analytes sont mesurés.
Cependant, cette approche est confrontée à la présence de nombreuses interférences spectrales venant des éléments de la matrice[36]. De plus, les
Prisme Détecteur bidimensionnel
refroidi par effet Peltier
17
échantillons chargés en sel dissout, après une mise en solution par fusion, par exemple, peuvent induire des effets matriciels, diminuant ainsi les performances analytiques de l’instrument. Il est possible d’en constater les effets à l’aide de l’équation de Saha [16] :
=(2 !"#$%) / '!ℎ 2)*+ )* , -. /01
Cette équation représente le ratio d’éléments ionisés versus les éléments non-ionisées ([X+/X]). Cette équation est une fonction de la densité du
nombre d’électrons (ne), de la température d’ionisation (Ti) et du therme , 23. 451
qui représente l’énergie d’ionisation de l’analyte. Finalement, k et h représentent respectivement les constantes de Boltzmann et de Plank alors de me équivaut à la masse d’un électron. Il est donc possible d’observer que
la densité du nombre d’électrons est inversement proportionnelle au taux d’ionisation d’un analyte donné. En d’autres mots, plus il y aura d’électrons à l’intérieur du plasma, moins il y aura d’atomes ionisés à une température donnée.
Une séparation des analytes de leur matrice et l’utilisation de références internes peuvent contrer ces effets et améliorer la qualité des résultats.
1.3.2 Spectroscopie d’émission atomique à plasma micro-ondes (MP-AES)
Le premier appareil à plasma micro-ondes (MP-AES) est arrivé sur le marché en 2011. Son principe de fonctionnement est similaire à celui du plasma à couplage d’argon inductif, à quelques différences près. En effet, son plasma n’est pas généré par un champ électrique, mais plutôt par un champ magnétique, ce qui génère un plasma très robuste capable de traiter une grande variété de matrices. Un guide d’ondes d’hyperfréquence concentre le champ magnétique autour de la torche. Un champ magnétique
18
axial et un champ électrique radial accumulent et focus l’énergie des micro-ondes afin de créer le plasma. Par la suite, le MP utilise le même principe d’analyse des photons suite à l’émission de photons par les électrons excités revenant à leur état fondamental.
Les micro-ondes produits par un magnétron, typiquement à 2,45 GHz, pénètrent la cavité où est situé le tube de quartz. Ils causent ainsi une oscillation des électrons présents dans le gaz plasmagène. Ces électrons oscillants entrent alors en collision avec les autres atomes présents dans le milieu et créent ainsi la haute température du plasma.
L’un des avantages principaux du MP comparativement à l’ICP est son coût d’utilisation [37]. En effet, le gaz plasmagène du MP, étant de l’azote, est moins dispendieux que celui utilisé par l’ICP, utilisant l’argon. De plus, il est possible pour les utilisateurs de MP d’utiliser un générateur d’azote purifiant ce gaz directement de l’air ambiant, diminuant ainsi grandement le coût d’utilisation à long terme [37]. Le MP utilise une faible quantité d’argon lors de l’ignition du plasma pour ensuite ne consommer que de l’azote pour la production du plasma [38]. Il est aussi possible d’utiliser un mélange d’oxygène/azote afin d’analyser des matrices organiques en ne modifiant que légèrement le système [38]–[40], comme par l’ajout d’un module externe de contrôle des gaz pour pallier aux effets de matrice dus à la température plus froide du plasma en comparaison à l’ICP. Ce module a pour tâche d’introduire de l’air dans le plasma afin de maintenir sa stabilité et minimiser le bruit de fond originant des composés organiques. L’oxygène de l’air réagit avec la matrice organique à haute température afin de former du monoxyde et dioxyde de carbone (CO et CO2 respectivement), ce qui
prévient les émissions du carbone et les dépôts sur la torche. Il est aussi possible d’ajouter un nébuliseur mélangeur de flux (Flow Blurring nebulizer (FBN)) dont le rôle est de mélanger la solution liquide et le gaz du nébuliseur afin de produire un aérosol supérieur ayant de particules plus fines en comparaison avec les nébuliseurs concentriques conventionnels [39].
19
Le plasma obtenu dépend du gaz plasmagène utilisé. En effet, un plasma à base d’air aura une couleur bleu-gris due aux liens NO à 200-290 nm et aux émissions des liens OH à 306 nm. Dans le cas du plasma à base d’azote, le plasma possède une couleur rosée. Cette couleur vient des raies spectrales intenses des liens N-N à 330-390 nm[38].
En plus de son détecteur multiélémentaire, le MP a aussi l’avantage de n’utiliser aucun gaz inflammable ou de gaz toxique, par exemple l’acétylène ou encore le peroxyde d’azote, comme c’est le cas pour le spectromètre d’absorption atomique à flamme (AASF). De plus, la température du plasma est largement supérieure à celle de la flamme du AASF, ce qui facilite l’analyse d’éléments réfractaires et contribue à diminuer l’effet de matrice [38].
L’une des principales limitations à l’utilisation du MP est lors de l’analyse de matrices ayant une charge élevée en sel. En effet, lorsque la charge totale en sel dissout est élevée, les performances analytiques de l’appareil en sont diminuées en plus d’augmenter les risques d’obstruction du nébuliseur ou de dévitrification de la torche [37]. Il est conseillé de diluer les échantillons surchargés, mais, se fessant, la concentration de l’analyte sera également réduite, limitant dans certaines occasions sa détection ou sa quantification. La température du plasma d’un MP peut aussi être considérée comme un obstacle lorsque cette dernière est comparée à celle d’un ICP. La température du MP atteint une température de 5000 K alors que celle du ICP est de 7500 K. Comme démontré à l’aide de l’équation de Saha [16], la température d’ionisation est directement proportionnelle au taux d’ionisation d’un analyte en particulier. Donc avec une température de plasma plus faible, moins d’atomes seront ionisés à l’intérieur du plasma. De plus, lors de l’introduction d’une matrice ayant une concentration élevée en sel dissout, l’ion métallique du fondant présent en grande quantité aura tendance à capturer un électron du plasma et former une espèce atomique,
20
ce qui aura pour effet de diminuer la densité d’électrons et par conséquent, la température du plasma. Ceci est particulièrement vrai dans le cas des éléments à faibles énergies d’ionisation comme le sodium.
C’est pour ces raisons qu’il est nécessaire de développer des outils afin de réduire la présence des ions non désirés de la matrice. En le faisant préalablement à l’introduction de l’échantillon dans le plasma, cela permet de diminuer les limites de détection et d’augmenter la précision des analyses pour un ou plusieurs métaux se trouvant dans une matrice dont la charge saline est très élevée.
1.3.3 Spectrométrie d’émission atomique à plasma à couplage inductif coupler à un spectromètre de masse en tandem (ICP-MS/MS)
La spectrométrie de masse (MS pour mass spectrometer) est certainement l’une des techniques analytiques ayant connu le plus d’innovations depuis les années 40. En effet, une multitude de domaines utilise cette technique afin d’analyser une multitude d’analytes ayant leurs propriétés propres, allant de l’analyse élémentaire aux analyses de biomolécules de plus en plus massives.
Dans le cas du ICP-MS/MS, le système d’introduction des échantillons est très similaire à celui du ICP-AES. La différence vient du fait qu’il ne sera plus question de mesurer les photons émis lors de la relaxation électronique des éléments, mais plutôt les analytes ionisés. En effet, une fois l’échantillon nébulisé et ionisé, un flux d’ions sera dirigé vers deux cônes constitués de nickel, qui font office de séparateur entre l’interface du plasma, qui est à pression atmosphérique, vers l’interface du spectromètre de masse qui est, pour sa part, sous vide. Le vide de l’ordre de 10-5 à 10-7 Torr est obtenu à
l’aide de pompes mécaniques et turbomoléculaires. Le flux d’ions est par la suite dévié afin de discriminer les particules non ionisées pour finalement entrer dans le premier quadripôle. Un quadripôle est composé de quatre
21
tiges dans lesquelles un courant électrique circule. De ce fait, il est possible de créer un champ magnétique et ainsi ségréger les analytes selon leurs ratios masse sur charge (m/z). Une fois le premier ratio m/z sélectionné, le flux d’analyte passe dans la chambre de collision-réaction. Ce dispositif sert à éliminer les interférences liées aux ratios m/z, qu’elles soient isobariques ou moléculaires.
Une interférence isobarique est observée lorsque l’analyte mesuré est en compétition avec un autre élément ayant le même ratio m/z. À titre d’exemple, il est possible d’observer ce type d’interférence lors de l’analyse du potassium-40 (40K) puisque le calcium (40Ca) possède aussi un isotope
de même ratio m/z. Dans le cas des interférences moléculaires, l’analyse du platine-195 (195Pt) est biaisée par une interférence moléculaire venant de
l’hafnium et de l’oxygène (179Hf16O). Pour éliminer ces interférences, il est
possible de faire circuler un gaz collisionnel, communément de l’hélium. Dans le cas des interférences isobariques, il est possible de jouer avec la chimie des analytes à l’intérieur du flux d’ion. Pour ce faire, des gaz réactionnels, communément de l’ammoniac (NH3), de l’oxygène (O2) ou
encore de l’hydrogène (H2), afin de changer le ratio m/z de l’analyte ou
encore de son interférent.
Avant d’atteindre le détecteur, le flux d’ion entre dans le deuxième quadripôle. Il est possible de changer la sélection du ratio m/z afin d’éliminer les interférences ayant été corrigées par la cellule de collision-réaction.
22
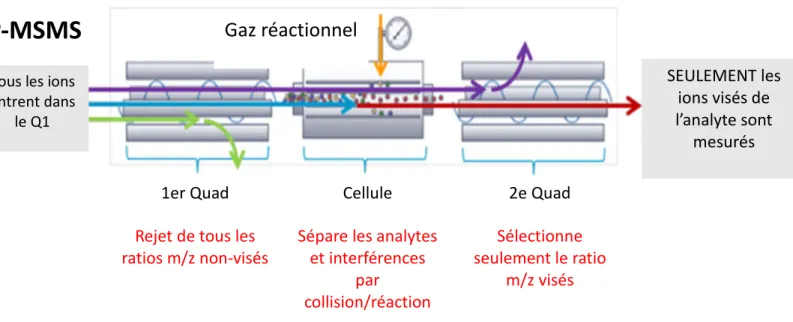
Figure 6 : Schéma simplifié du trajet parcouru par les ions dans un spectromètre de masse avec une cellule de collision-réaction. [41]
Finalement, le faisceau d’ions termine sa course vers le détecteur de l’appareil. Il existe plusieurs types de détecteurs liés au ICP-MS/MS. Dans le cadre de ce projet, l’ICP-MS utilisait comme détecteur un tube photomultiplicateur (PMT). Les PMT utilisent l’effet photoélectrique comme principe de base. Ce phénomène a été compris pour la première fois par Hertz en 1887, puis mis en théorie par Einstein en 1905. L’effet photoélectrique peut être défini comme étant l’émission d’électrons par un matériau lorsque ce dernier est soumis à une radiation électromagnétique d’énergie suffisamment élevée, ce paramètre dépendant du matériau lui-même. Les PMT remplacent les phototubes qui sont aujourd’hui désuets. Le principe de fonctionnement d’un PMT est le suivant ; les ions positifs frappent un convecteur (cristal ou dynode) et les photons ou les électrons produits viennent frapper une cathode qui produit des électrons. Le nombre d’électrons produit dépend du type de particules primaires, de son énergie et de la surface incidente.
ICP-MSMS
Tous les ions entrent dans
le Q1
Gaz réactionnel
1er Quad
Rejet de tous les ratios m/z non-visés
Cellule
Sépare les analytes et interférences par collision/réaction 2e Quad Sélectionne seulement le ratio m/z visés SEULEMENT les ions visés de l’analyte sont mesurés
23 1.4 Inconvénients de la fusion
La section 1.2 a précédemment présenté plusieurs avantages liés à la digestion par fusion. Cette technique n’est cependant pas sans lacunes. Pour commencer, les méthodes de digestion impliquant une fusion demandent d’ajouter une grande quantité de fondant en rapport avec la quantité d’échantillons. Le lithium et le sodium sont des éléments facilement ionisables et libèrent donc facilement un électron lors d’une excitation thermique. Comme discuté précédemment à l’aide de l’équation de Saha, il est démontré que l’augmentation de la densité du nombre d’électrons vient à diminuer le ratio d’ionisation d’un élément donné [42]. De plus, la possible présence de contaminants à l’intérieur même des fondants peut aussi influencer les paramètres analytiques des méthodes d’analyses, en particulier lors d’analyse de métaux traces. Il existe, par contre, sur le marché des fondants de grades ultrapures diminuant ainsi l’influence de cette contamination sur les échantillons. Ce sujet sera d’ailleurs étudié dans le cadre de ces travaux. Par la suite, il est évidemment impossible de doser les analytes constituants des fondants comme le sodium dans le cas du Na2O2 ou encore le calcium lors de l’utilisation de CaCO3. Afin de pallier à
ce problème, l’utilisation stratégique d’un autre fondant peut s’avérer nécessaire. Aussi, les fortes charges en sels engendrées par la fusion alcaline sont souvent incompatibles avec l’instrumentation utilisée pour l’analyse élémentaire. Cet inconvénient a des répercussions autant du point de vue analytique qu’instrumental. Une piste de solution se trouve dans la possibilité de diluer l’échantillon fusionné, mais ce facteur peut souvent être de l’ordre de 5000 et, lors de l’analyse de faibles quantités de métaux, il est possible de se retrouver sous les limites de détection des méthodes analytiques [43]. Finalement, comme le mode de détection en spectroscopie d’absorption atomique est optique, le choix des longueurs d’onde à utiliser est très important afin de limiter la présence d’interférences spectrales. Le groupe de Balaram [4] s’est d’ailleurs penché sur ce sujet et a étudié les interférences de plusieurs métaux d’intérêts à l’aide du MP-AES.
24
La mise en solution par fusion au Na2O2 est une technique efficace
possédant plusieurs avantages, comme mentionné dans les sections précédentes. Elle est cependant accaparée de quelques défauts pouvant être éliminés à l’aide d’une méthode visant à réduire, voire même, à retirer les éléments responsables des inconvénients liés à ce type de préparation d’échantillon. Il est donc important d’élaborer et de développer une méthode de séparation du sodium d’une matrice géologique après une mise en solution par fusion à l’aide du Na2O2 comme fondant. Il sera d’ailleurs
question des multiples techniques de séparation disponibles dans la prochaine section de ce mémoire.
1.5 Techniques de séparation
Que ce soit pour l’analyse de composés organiques ou encore de métaux, une séparation chromatographique est très souvent nécessaire en chimie analytique afin d’obtenir des résultats justes et précis. Il existe plusieurs techniques reliées à la séparation de diverses espèces chimiques et ce, dans autant de matrices différentes. Cette section fera un survol des approches disponibles dans le contexte d’analyse de métaux.
25
Figure 7 : Schéma des différentes techniques de séparation [44]
1.5.1 Extraction liquide (ELL) et extraction liquide-liquide sur phase solide
En premier lieu, l’extraction liquide-liquide (ELL) et l’extraction liquide-liquide ionique (ELLI) sont des techniques de séparation utilisées couramment dans l’industrie minière, notamment dans l’extraction et la séparation de terres rares et la purification de l’uranium [45]. La première technique consiste à utiliser une phase aqueuse et une phase organique hydrophobe auxquels il est possible d’ajouter des ligands spécifiques aux éléments à isoler. Cette technique est utilisée lors de l’extraction et la purification des métaux de terres rares, soit les lanthanides[46]. Concernant l’extraction liquide-liquide ionique, les ligands sont ajoutés dans un sel organique ayant une température de fusion relativement faible afin d’être liquide lors de l’extraction et de la manipulation. Cette technique est utilisée afin d’extraire des métaux de transition [47] ou encore des éléments du
26
groupe de platine [48]. Cependant, ce type d’extraction génère des quantités non-négligeable de solvant, déchets acides et radioactifs [46], [49] et c’est pourquoi cette technique ne sera pas explorée dans ce présent mémoire.
En second lieu, l’extraction liquide-liquide sur support solide (ELLS) consiste en un support de polymère poreux sur lequel est imprégné un ligand sélectif à un ou plusieurs éléments. La résine UTEVA d’Eichrom Technologies Inc, utilisée dans la séparation de l’uranium et du thorium[31] ou encore du neptunium [32], est un exemple de résine utilisant les concepts de l’ELLS. De plus, Larivière et coll.[27,30] ont travaillé sur la fonctionnalisation de nanomatériaux à base de carbone et de silice afin d’extraire les lanthanides. Comme les ligands servant à extraire les métaux des échantillons sont imprégnés sur le support solide, cette technique de séparation est beaucoup plus verte que l’extraction liquide-liquide. Cette technique a aussi la possibilité d’être sélective à un élément ou encore à une famille d’éléments comme les lanthanides. Cependant, comme les ligands ne sont pas greffés sur le support solide, la durée de vie de la résine est souvent inférieure aux résines dont les groupements fonctionnels sont chimiquement liés au support. En effet, il peut se produire des modifications ou encore la perte du ligand lors des utilisations. De plus, comme le développement de la technique date de la fin des années 90 [50], il s’agit d’une technique récente et les résines commerciales peuvent être plus dispendieuses que les techniques ayant fait leurs preuves depuis des décennies.
1.5.2 Chromatographie d’échange ionique (EI)
Troisièmement, la chromatographie d’échange ionique (EI), est une technique très prometteuse qui a permis le développement de plusieurs méthodes chromatographiques, et ce, avec une multitude de matrices distinctes[21], [36], [51]–[54]. En effet, l’utilisation de résine échangeuse d’ions est une stratégie communément utilisée dans la pré-concentration et l’élimination de constituant de la matrice dans des échantillons aqueux [36].
27
Une résine est composée de petites billes de polymère poreuses, souvent en polystyrène (PS) ou encore du divinylbenzène (DVB) auxquelles est greffé des groupements fonctionnels. Il est possible de diviser les résines d’échange ionique en deux grandes familles, soit les résines anioniques et les résines cationiques. Pour la première, les molécules attachées sont de types acides, par exemple des groupements amines tertiaires, alors que celles attachées sur les résines cationiques sont de types basiques, par exemple des groupements sulfonates. Par conséquent, les résines anioniques auront une affinité avec les anions tandis que les cations auront des affinités avec les résines cationiques. Lorsqu’un échantillon, nommé phase mobile, est en contact avec la résine, les ions de ce dernier se diffusent de la phase mobile vers la phase stationnaire, la résine, dépendamment de leurs affinités avec cette dernière. C’est ainsi que peut s’effectuer la séparation des ions de l’échantillon. Dans le cas d’une résine anionique, il est possible d’illustrer ce phénomène à l’aide de l’équation suivante :
6 + 7 8 ↔ 7 6 + 8
Dans cette équation, A- représente un ion négativement chargé, R+ le
groupement fonctionnel attaché à la résine anionique et Y- le contre-ion, soit
Cl- ou OH-, dépendamment des résines. Il est alors possible de déterminer
le coefficient de distribution (Kd) entre la résine et l’analyte avec l’équation
suivante :
:
;=
<é $>! ? @A$?>Dans celle-ci, Crésine correspond à la concentration de l’analyte retenue sur
la résine alors que Csolution désigne pour sa part la concentration de ce même
28
Plusieurs méthodes d’analyses ont été développées à l’aide de ces types de résine afin d’extraire des PGE [9], [10], [36], [51], [53], [55]. Comme mentionné dans les chapitres précédents, le sodium provenant du fondant (Na2O2) doit être séparé d’une matrice minérale suite à une mise en solution
par fusion. Étant donné le ratio fondant/échantillon très en faveur du numérateur, l’utilisation d’une résine ayant une affinité nulle pour le sodium serait envisageable. Le choix s’est donc tourné vers les résines de type anionique de la compagnie Dow Chemical, plus précisément la Dowex 1x8. En effet, la résine Dowex 1x8 est la résine anionique utilisée pour le développement de la présente méthode grâce à sa grande disponibilité, son faible coût et sa capacité à capter des anions. Le cation de sodium n’a aucune affinité avec ce type de résine, ce qui empêche une saturation rapide de cette dernière. Comme il est démontré sur la figure 8, la Dowex 1x8 est formée d’une amine tertiaire retenue sur un copolymère de PS et de DVB.
Figure 8 : Structure de la résine Dowex 1x8
1.5.3 Extraction sur phase solide (SPE)
Un autre type de séparation, plus récente, est la chromatographie d’extraction sur phase solide, ou solide phase extraction (SPE). Il est possible de sous-diviser la SPE en quatre familles, dépendamment de la nature du support solide lui-même, comme le montre le schéma 7. Il sera cependant
29
question, dans ce mémoire, que de la famille dont le support solide est organique.
Contrairement à la chromatographie d’échange ionique, il s’agit d’une technique pouvant être très sélective. En effet, des groupements fonctionnels greffés sur la phase stationnaire peuvent complexer un élément en particulier. Elle est couramment utilisée lors de la séparation de composés dans des matrices complexes, comme le sang humain. Boatto et coll.[56] utilisent cette technique afin de séparer et analyser un dérivé d’amphétamine dans l’urine. Mais cette technique possède aussi la capacité de séparer les métaux comme l’ont démontré Pai et coll [57], dont l’objectif était de séparer des métaux lourds d’eau de mer à l’aide de la SPE. En effet, ils ont utilisé la résine Chelex-100 (Bio-Rad, USA) à un pH entre 6,0 et 7,0 afin d’être en mesure de ségréger les métaux comme le plomb, le cadmium, le chrome, le fer, le manganèse, le cobalt et le cuivre, pour n’en nommer que quelques-uns, d’une matrice saline. Le but était de trouver le pH idéal afin de retenir les métaux d’intérêts sur la résine et d’éliminer le calcium et le magnésium des échantillons d’eau salée puisque ces derniers entraient en compétition avec les métaux lourds sur la résine Chelex-100. Cette problématique n’est observable qu’avec ce type de matrice, car, toujours selon l’auteur, il est possible de complètement éliminer le Cu(II), le Fe (III) et le Cr(III) à un pH aussi bas que 2,5, dans un échantillon d’eau douce. Une autre résine a aussi été étudiée dans le cadre de ce projet. Il s’agit d’une résine chélatante utilisant la technique SPE. La résine Nobias PA-1 de Hitachi High-Technology a été utilisée dans plusieurs articles scientifiques [26], [58]–[61] et a permis l’analyse de métaux comme le zinc, le cuivre, l’aluminium, le plomb et le fer et autres jusqu’aux éléments de terres rares à l’état de trace dans de l’eau de mer. Il est à noter que l’eau de mer a une salinité moyenne de 35 g/L dont 10 g/L en sodium. Selon les articles présentés précédemment[26], [58]–[61], les familles des alcalins et alcalino-terreux ne seraient pas adsorbés sur la résine Nobais PA-1 et c’est pourquoi
![Figure 1 : Valeur de la production minérale au Canada de 1999 à 2016 par catégorie (en milliards de dollars canadiens) [2]](https://thumb-eu.123doks.com/thumbv2/123doknet/3459274.101036/13.918.127.792.370.803/figure-valeur-production-minérale-canada-catégorie-dollars-canadiens.webp)