Université de Sherbrooke
Développement et applications d’un outil bio-informatique pour la détection de similarités de champs d’interaction moléculaire.
Par
Matthieu Chartier Programme de biochimie
Thèse présenté(e) à la Faculté de médecine et des sciences de la santé en vue de l’obtention du grade de philosophiae doctor (Ph.D.)
en biochimie
Sherbrooke, Québec, Canada Mai 2016
Membres du jury d’évaluation
Pr. Rafael Najmanovich, Département de Biochimie, Université de Sherbrooke Pr. François Bachand, Département de Biochimie, Université de Sherbrooke
Pr. Pierre Lavigne, Département de Biochimie, Université de Sherbrooke
Pr. Éric Marsault, Département de Pharmacologie-physiologie, Université de Sherbrooke Pr. Guillaume Lamoureux, Département de Chimie et Biochimie, Université Concordia
Around here, however, we don't look backwards for very long. We keep moving forward, opening up new doors and doing new things, because we're curious...and curiosity keeps leading us down new paths. - Walt Disney
Développement et applications d’un outil bio-informatique pour la détection de similarités de champs d’interaction moléculaire.
Par
Matthieu Chartier Programme de biochimie
Thèse présentée à la Faculté de médecine et des sciences de la santé en vue de l’obtention du grade de philosophiae doctor (Ph.D.) en biochimie. Faculté de médecine et des sciences de la santé, Université de Sherbrooke, Sherbrooke,
Québec, Canada, J1H 5N4
Les méthodes de détection de similarités de sites de liaison servent entre autres à la prédiction de fonction et à la prédiction de cibles croisées. Ces méthodes peuvent aider à prévenir les effets secondaires, suggérer le repositionnement de médicament existants, identifier des cibles polypharmacologiques et des remplacements bio-isostériques. La plupart des méthodes utilisent des représentations basées sur les atomes, même si les champs d’interaction moléculaire (MIFs) représentent plus directement ce qui cherche à être identifié.
Nous avons développé une méthode bio-informatique, IsoMif, qui détecte les similarités de MIF entre différents sites de liaisons et qui ne nécessite aucun alignement de séquence ou de structure. Sa performance a été comparée à d’autres méthodes avec des bancs d’essais, ce qui n’a jamais été fait pour une méthode basée sur les MIFs. IsoMif performe mieux en moyenne et est plus robuste. Nous avons noté des limites intrinsèques à la méthodologie et d’autres qui proviennent de la nature. L’impact de choix de conception sur la performance est discuté.
Nous avons développé une interface en ligne qui permet la détection de similarités entre une protéine et différents ensembles de MIFs précalculés ou à des MIFs choisis par l’utilisateur. Des sessions PyMOL peuvent être téléchargées afin de visualiser les similarités identifiées pour différentes interactions intermoléculaires.
Nous avons appliqué IsoMif pour identifier des cibles croisées potentielles de drogues lors d’une analyse à large échelle (5,6 millions de comparaisons). Des simulations d’arrimage moléculaire ont également été effectuées pour les prédictions significatives. L’objectif est de générer des hypothèses de repositionnement et de mécanismes d’effets secondaires observés. Plusieurs exemples sont présentés à cet égard.
Mots clés : Champs d’interaction moléculaire, détection de similarités, réactivité croisée, reconnaissance moléculaire, polypharmacologie, effets secondaires, repositionnement de médicaments.
S
UMMARYDevelopment and applications of a bioinformatic tool to detect molecular interaction field similarities.
By
Matthieu Chartier Biochemistry Program
Thesis presented at the Faculty of medicine and health sciences for the obtention of Doctor degree diploma philosophiae doctor (Ph.D.) in Biochemistry.
Faculty of medicine and health sciences, Université de Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4
Methods that detect binding site similarities between proteins serve for the prediction of function and the identification of potential off-targets. These methods can help prevent side-effects, suggest drug repurposing and polypharmacological strategies and suggest bio-isosteric replacements. Most methods use atom-based representations despite the fact that molecular interaction fields (MIFs) represents more closely the nature of what is meant to be identified.
We developped a computational algorithm, IsoMif, that detects MIF similarities between binding sites. We benchmark IsoMif to other methods which has not been previously done for a MIF-based method. IsoMif performed best in average and more consistently accross datasets. We highlight limitations intrinsic to the methodology or to nature. The impact of design choices on performance is discussed.
We built a freely available web interface that allows the detection of similarities between a protein and pre-calculated MIFs or user defined MIFs. PyMOL sessions can be downloaded to visualize similarities for the different intermolecular interactions.
IsoMif was applied for a large-scale analysis (5,6 millions of comparisons) to predict off-targets of drugs. Docking simulations of the drugs in the binding site of their top hits were performed. The primary objective is to generate hypotheses that can be further investigated and validated regarding drug repurposing opportunities and side-effect mechanisms.
Keywords : Molecular interaction fields, detection of similarities, cross-reactivity, molecular recognition, polypharmacology, side-effects, drug repurposing.
Résumé ... iv
Summary ... v
Table des matières ... vi
Liste des figures ... x
Liste des tableaux ... xii
Liste des abréviations ... xiii
Chapitre 1 - Introduction ... 1
Fonctions des protéines ... 1
Déterminants de la fonction moléculaire ... 2
Détermination de la fonction moléculaire via la séquence et la structure ... 3
Limites de similarités de séquence et de structure ... 5
Choix de la méthode et contextes d’utilisation ... 5
Méthodes de similarités de site de liaison ... 7
Problématique et hypothèses ... 14
Représenter la fonction moléculaire ... 14
Champs d’interaction moléculaire ... 15
Objectifs ... 16
Objectif #1 – Développement d’une nouvelle méthode (Chapitre 2, article 1) ... 17
Objectif #2 – Validation et comparaison des performances (Chapitre 2, article 1) ... 18
Objectif #3 - Développement d’une interface en ligne (Chapitre 3, article 2) ... 19
Objectif #4 – Application à large échelle (Chapitre 4) ... 19
Chapitre 2 - Article 1 ... 20
Abstract ... 21
Introduction... 22
Methods ... 26
Molecular Interaction Fields ... 26
Search and Scoring of the MIF Similarities ... 30
Data Sets and Performance Evaluation ... 32
Visualization of MIF Similarities ... 34
Results ... 35
Validation with Homogenous, Steroid, and SOIPPA Data Sets ... 38
Mean AUC Values ... 40
Enrichment Factors ... 41
Extended Validation with the PDBbind and sc-PDB Data Sets ... 42
Effect of Binding Site Definition ... 43
Effect of Structural Variability ... 45
Examples and Applications ... 47
Discussion ... 51 Conclusion ... 55 Acknowledgment ... 56 Chapitre 3 - Article 2 ... 57 Abstract ... 58 Summary ... 58
Availability and Implementation: ... 58
Contact: ... 58
Supplementary information: ... 58
Background ... 58
Submitting a job ... 59
Cropping the cavities ... 59
Results Page ... 60
Example Application ... 60
Acknowledgments ... 62
Chapitre 4 - Détection à grande échelle de cibles croisées : repositionnement de médicaments et effets secondaires ... 63
Introduction... 63
Matériel et méthodes ... 66
Sites de liaison ... 66
Drugs ... 66
Pisces ... 66
Calculs des similarités et arrimage moléculaire ... 67
Compilation d’effets secondaires et de données connexes ... 67
Analyses et disponibilité des résultats ... 68
Résultats ... 69
Effets secondaires ... 70
Similarités de site de liaison et arrimage moléculaire ... 70
Listes de cas intéressants ... 70
Discussion ... 72
Ensembles de données ... 72
Promiscuité des ligands ... 73
Promiscuité des cibles ... 75
Hypothèses de repositionnement ... 77
Acetazolmaide et Chorismate pyruvate lyase ... 77
Raloxifène et CviR ... 78
Hypothèses de mécanismes d’effets secondaires ... 82
Zanamivir ... 82
Captopril ... 83
Retour sur l’approche et les hypothèses ... 86
Conclusion ... 87
Chapitre 5 - Discussion générale ... 88
Limites de la méthode et perspectives d’amélioration ... 88
Détection de cavités ... 88
Flexibilité des protéines ... 92
Représentation avec les champs d’interaction moléculaire ... 94
Algorithme de recherche ... 99
Fonction de pointage ... 103
Évaluation de la performance ... 105
Jeux de données ... 105
Définition de vrais positifs ... 106
Définition du site de liaison ... 107
Difficultés générales et limites de la nature ... 109
Compromis de sensibilité ... 109
Modes de liaison ... 110
Multiples fonctions et sous-similarités ... 111
Applications et interface en ligne ... 111
Liste des références ... 114
Annexes ... 125
Annexe B – Données supplémentaires du chapitre 3 (article 2) ... 143 Annexe C – Données supplémentaires du chapitre 4... 147
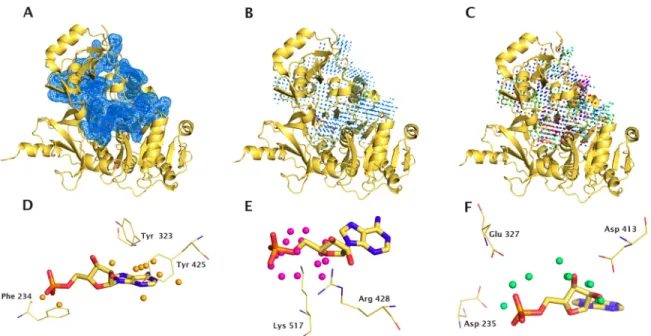
Figure 2.1 – MIF representation for gramicidin synthetase 1 bound to AMP ... 30
Figure 2.2 – Boxplot of the performance for the Kahraman dataset ... 37
Figure 2.3 – Boxplot of the performance for the Homogenous dataset ... 38
Figure 2.4 – Boxplot of the performance for the Soippa dataset ... 40
Figure 2.5 – Performance with different binding site definitions ... 45
Figure 2.6 – Similarities between 1E8X and 1DV2 ... 48
Figure 2.7 – Similarities between 1AHB and 3GEY ... 51
Figure 3.1 – Similarities between 6COX and 1RJ6 ... 61
Figure 4.1 – Fraction des MIFs pour toutes les entrées Pisces et pour les plus communes . 71 Figure 4.2 – Structure et cavités de la prostaglandine-endoperoxide synthase ... 74
Figure 4.3 – MIF du récepteur des minéralocorticoïdes (4PF3_2) ... 76
Figure 4.4 – Similarités entre l’anhydrase carbonique et la chorismate pyruvate-lyase ... 78
Figure 4.5 – Superposition et similarités du récepteur à estrogène et du récepteur CviR .... 80
Figure 4.6 – Similarités entre le récepteur à estrogène et le récepteur CviR ... 81
Figure 4.7 – Similarités entre la neuraminidase liée au zanamivir et le canal potassique voltage dépendant 3HFE_1 ... 83
Figure 4.8 – Entrée 2X8Z_X8Z_1615_A_- dans l’interface en ligne ... 84
Figure 4.9 – Cible croisée 1FV1_7 identifiée avec le site 2X8Z_X8Z_1615_A_- ... 85
Figure 5.1 – Cavités détectées par GetCleft avec différents rayons de sphères ... 90
Figure 5.2 – Cavité trouvée au site de liaison à l’ATP pour les conformations active et inactive de p38𝛼𝛼 ... 91
Figure 5.3 – Distances entre paires de sondes, sommets et arêtes du graphe ... 93
Figure 5.4 – Similarités pour 3 sondes entre 6COX et 1RJ6 ... 95
Figure 5.5 – Sondes donneur et représentation du seuil d’angle limite ... 97
Figure 5.6 – Sondes aromatiques et représentation du seuil d’angle limite ... 99
Figure 5.7 – Illustration comparative des différentes résolutions de la grille ... 99
Figure 5.8 – Comparaison de la taille du FAD et de l’ATP ... 102
Figure 5.9 – Seuils de définition de sites de liaison ... 109
Figure 5.10 – Similarités entre 6COX et la phospholipase A2 et le récepteur à l’androgène ... 113
Figure A.2 – Angle vectors and reference atoms ... 139
Figure A.3 – Graph representation of the probe thresholds ... 140
Figure A.4 – Association graph example ... 141
Figure A.5 – Top hits of 1BXM ... 142
Figure B.1 – Cropping the cavities ... 143
Figure B.2 – Visualisation of the user defined target cavities ... 144
Figure B.3 – IsoMif Finder results page example ... 145
Figure B.4 – PyMOL session showing the similarities ... 146
Figure C.1 – Sites de liaison de la rhodopsin II ... 195
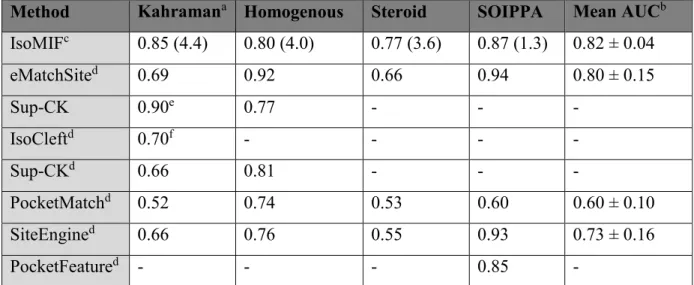
Table 2.1 – AUCs for IsoMIF and 6 other methods on different datasets ... 36
Table 4.1 – Ligands prédits pour le récepteur des minéralocorticoïdes (4PF3_2) ... 76
Table 4.2 – Sites de liaison de Drugs liés au raloxifène similaires à 3QP4_1 ... 79
Table A.1 – IsoMif Interaction Matrix ... 125
Table A.2 – Probe thresholds ... 125
Table A.3 – Amino acid atom types and reference atoms for angle calculations ... 125
Table A.4 – Performance for Bron & Kerbosch search thresholds ... 128
Table A.5 – Performance for the PDBbind and scPDB datasets ... 129
Table A.6 – Average AUC on perturbed structures ... 129
Table A.7 – Best AUC using perturbed structures ... 130
Table A.8 – Top 10 hits found for query 1BXM sorted by MSSa ... 130
Table A.9 – Performance with different geometric distance thresholds ... 131
Table A.10 – AUCs for Kahraman excluding PO4 entries ... 131
Table A.11 – AUCs for Kahraman including PO4 entries ... 132
Table A.12 – AUCs for homogenous dataset ... 133
Table A.13 – AUCs for the Steroid dataset ... 135
Table A.14 – AUCs for the Soippa dataset ... 135
Table C.1 – Liste non redondante des ligands de l’ensemble Drugs ... 147
Table C.2 – Liste des 400 entrées de l’ensemble Drugs ordonnés par nombre de cibles ... 153
Table C.3 – Cibles prédites plusieurs fois pour la même drogue ... 164
MIF CF PDB RM RMSD Zx
Champ d’interaction moléculaire
Molecular Interaction Field
Fonction de complémentarité
Complementarity Function Protein Data Bank
Récepteur des minéralocorticoïdes
Mineralocorticoïd Receptor Root-Mean-Square Deviation
Valeur centrée réduite
Fonctions des protéines
Les protéines sont diverses en forme et en fonction. Leur concentration varie d’un tissu à l’autre et dans le temps. Elles jouent des rôles essentiels dans les processus métaboliques et leur dérégulation ou des mutations à des positions importantes sont souvent la cause de plusieurs maladies. Une connaissance approfondie de la fonction d’une protéine permet une bonne compréhension de son rôle au sein de la cellule et de l’organisme et s’avère nécessaire lors de l’élaboration d’une approche thérapeutique efficace. La fonction d’une protéine peut désigner une réaction enzymatique ou peut prendre un sens plus global, soit à l’échelle de l’organisme ou au niveau cellulaire, par exemple le transport, la mitose ou la motilité cellulaire. À plus petite échelle, la fonction moléculaire peut désigner l’ensemble des partenaires d’interaction capables de lier une protéine tels une petite molécule, un cofacteur, un agent thérapeutique ou des substrats de taille plus large comme un peptide ou un brin d’ADN.
Les interactions protéine-protéine et protéine-acides nucléiques sont ubiquitaires et essentielles aux fonctions cellulaires. Cependant, les interactions protéine-ligand sont l’objet principal de cette thèse. Il est possible de prédire in silico la fonction moléculaire d’une protéine en utilisant des principes thermodynamiques et des modèles paramétrés à l’aide de données expérimentales. La fonction moléculaire dans cette thèse réfère aux petites molécules capables de lier la protéine avec affinité. Ces techniques, par exemple le criblage virtuel à haut débit dans le cas spécifique des petites molécules, ont une marge d’erreur qui provient principalement de l’imperfection des fonctions de pointage, mais demeurent utiles pour identifier et optimiser des composés lors du développement d’inhibiteurs (Kitchen et al., 2004). La détermination ab initio de la fonction moléculaire à partir de la séquence, c’est à dire de connaître la fonction d’une protéine strictement à partir de premiers principes et sans aucune utilisation de données externes est un défi d’un autre ordre qui demeure pour l’instant à accomplir. La détermination ab initio de la structure
tertiaire d’une protéine à partir de la séquence est une partie du problème et est un domaine de recherche qui a beaucoup progressé (Dill et al., 2008). Actuellement, d’un point de vue bio-informatique, le mieux que l’on puisse faire pour déterminer la fonction moléculaire d’une protéine d’intérêt est d’utiliser les informations d’autres protéines comme leur séquence, leur structure et leur profil de liaison, combinés à des méthodes de simulation d’arrimage, d’homologie de structure, et de détection de similarités et d’alignement.
Déterminants de la fonction moléculaire
Même si la prédiction ab initio n’est pas actuellement possible, il y a tout de même une bonne compréhension des déterminants de la reconnaissance moléculaire c’est-à-dire des facteurs qui régissent les interactions intermoléculaires. D’abord, la forme et le volume du site de liaison permettent une complémentarité stérique entre la protéine et ses partenaires d’interaction. La forme et le volume du site de liaison peuvent, dans certains cas, permettre de discriminer quels ligands peuvent s’y lier (Kahraman et al., 2007).
Cependant, les protéines sont des entités dynamiques et la forme d’un site de liaison peut varier. Les propriétés thermodynamiques de la protéine dictent les mouvements accessibles à différentes échelles, notamment la réorganisation des chaînes latérales (Gaudreault et al., 2012a) ou les mouvements plus larges au niveau du squelette peptidique et des domaines (Chothia et al., 1983). Ces propriétés sont également déterminantes de la fonction moléculaire. La capacité à atteindre différents états structuraux, permet certains mécanismes importants dans les processus de reconnaissance moléculaire (Mittag et al., 2010) comme la modulation de l’activité par la liaison d’un ligand allostérique. Un exemple est celui de la modulation des interactions dans la portion cytosolique d’un GPCR par des agonistes ou antagonistes qui se lient dans la pochette extracellulaire du récepteur (Kenakin et Miller, 2010).
Lors de la liaison d’un ligand, il y a une perte ou un gain d’entropie conformationnelle de la protéine et du ligand, qui est parfois accompagné par un gain ou une perte enthalpique. Cette perte ou ce gain enthalpique provient de la perte ou du gain d’interactions entre le ligand et les molécules du solvant et des acides aminés au site de liaison de la protéine. La disposition de ces acides aminés est donc un autre facteur déterminant de la fonction moléculaire. Le ligand peut interagir non seulement avec les acides aminés du site de liaison, mais avec des cofacteurs, des molécules d’eau ou des ions métalliques via des interactions intermoléculaires telles que les interactions électrostatiques, les interactions de type van der Waals, les interactions Pi, les ponts hydrogènes. L’effet hydrophobe, c’est-à-dire la tendance qu’ont les surfaces hydrophobes du ligand à s’agréger avec celles de la protéine, du moins à réduire la surface en contact avec le solvant polaire, favorise l’interaction via un gain entropique de désolvatation. Ultimement, ce n’est pas l’identité des résidus qui est conservée, mais leurs propriétés physico-chimiques et les types d’interaction dans lesquels ils s’engagent. Par exemple, les kinases ont des résidus conservés dans la charnière, une région qui relie les lobes C et N-terminal du domaine catalytique. Bien que l’identité de ces résidus et leur nombre varient, la capacité de former des ponts hydrogènes à cet endroit avec le groupement adénine de la molécule d’ATP doit être conservée et cette propriété est exploitée lors du développement d’inhibiteurs (Xing et al., 2015). Les acides-aminés conservés dans un site de liaison ne sont pas nécessairement impliqués directement dans l’interaction avec les substrats, mais pourraient jouer un rôle tout aussi important pour la fonction moléculaire, comme la formation de ponts hydrogènes intraprotéiques qui stabilisent la structure de la forme liée ou pour prévenir la liaison de ligands compétitifs présents dans l’environnement de la protéine (Najmanovich et al., 2008).
Prédiction de la fonction moléculaire via la séquence et la structure tertaire
Outre les facteurs extrinsèques comme le pH capable de changer l’état de protonation de résidus importants ou les partenaires d’interactions capables de moduler les structures, les facteurs intrinsèques discutés dans la section précédente qui donnent à une protéine sa fonction moléculaire sont essentiellement encodés dans la séquence (structure primaire). En effet, la composition de la séquence globale confère la structure tertiaire et les propriétés
thermodynamiques (Frappier et Najmanovich, 2015) ainsi que la forme des cavités. Par rapport à un ancêtre commun, les séquences des protéines homologues tendent à diverger dans leur taxon au cours de l’évolution, mais les acides-aminés responsables des interactions déterminantes de la fonction moléculaire, directement ou indirectement, sont conservés au fil des générations suite aux pressions évolutives tant et aussi longtemps que cette fonction moléculaire confère aux individus de la population un avantage sélectif. Ainsi, les algorithmes de similarité de séquence tels FASTA (Pearson et Lipman, 1988) ou BLAST (Mount, 2007) permettent de retrouver des protéines homologues et d’inférer une fonction à une protéine d’intérêt en consultant les informations cataloguées des protéines homologues identifiées.
La base de données Uniprot et son interface web (Consortium, 2014) permettent de trouver des protéines notamment par mots clés ou via un alignement de séquence. Pour chaque protéine de la base de données, des informations en provenance d’autres bases de données connexes sont cataloguées, comme KEGG (Kanehisa et al., 2016) pour les voies métabolique, Gene Ontology (Gene Ontology Consortium et al., 2013) pour les fonctions biologiques, cellulaires et la localisation ou ChEMBL (Bento et al., 2014) pour l’activité biologique de petites molécules. ChEMBL est une ressource particulièrement intéressante, car elle permet d’obtenir un profil de liaison non seulement de molécules endogènes à l’être humain, mais de molécules issues de programmes de chimie médicinale. Pour une protéine étudiée en tant que cible primaire d’une maladie pour laquelle un inhibiteur est en développement, ces petites molécules capables de moduler l’activité des protéines homologues sont un bon point de départ pour le développement d’un inhibiteur.
Parfois la similarité de séquence, globale ou locale, est trop faible entre deux séquences homologues pour suggérer une proximité évolutive, mais la structure de la protéine et sa fonction moléculaire sont conservées. Au-delà de 40% d’identité de séquence, la structure est généralement conservée, mais une zone grise existe entre 20-35% d’identité (Rost, 1999) où la structure peut ne plus être conservée. Afin de complémenter les analyses de
similarités de séquence, les algorithmes de similarité de structure tel que DALI (Holm et Rosenström, 2010) ou FATCAT (Ye et Godzik, 2003) peuvent retrouver des protéines homologues du point de vue de la structure et ainsi, par référencement croisé, permettre d’obtenir des informations sur la fonction d’une protéine d’intérêt.
Limites de similarités de séquence et de structure
Dans le cas de la fonction moléculaire des interactions protéine-ligand, il est possible que deux protéines ne partagent aucune similarité de séquence ou de structure, mais aient la capacité de lier des substrats similaires (Chalk et al., 2004). Cela est possible si deux protéines divergent d’un ancêtre commun jusqu’à ce qu’il n’y ait plus de similarité de séquence significative et que la structure tertiaire d’une des protéines se modifie suite à des évènements évolutifs tels que des insertions ou délétions de domaines (Fong et al., 2007) sans affecter l’environnement physico-chimique du site de liaison ni la fonction moléculaire. C’est également possible si des pressions environnementales similaires s’exercent sur deux protéines non apparentées, les poussant à acquérir, un environnement physico-chimique similaire dans une cavité et donc une même fonction moléculaire. Les protéases sont un exemple de cette évolution convergente (Buller et Townsend, 2013). Dans ces deux scénarios d’évolution divergente et convergente, si la similarité de séquence ou de structure ne peut pas être utilisée, comment prédire les environnements physico-chimiques similaires susceptibles d’engendrer des similarités de profil de liaison et de fonction moléculaire?
Choix de la méthode de prédiction et contextes d’utilisation
Ultimement, la méthode à préconiser dépend du contexte et de l’objectif de recherche. Si un motif, par exemple une séquence signal, est recherché, les méthodes de détection de similarités de séquence locales sont idéales. Si l’objectif est l’identification de domaines structuraux, les méthodes de détection de similarités structurales globales sont plus efficaces. Si l’objectif est d’obtenir une classification hiérarchique pour un ensemble de
protéines, les méthodes de détection de similarités de séquence globale sont rapides et capables d’estimer la proximité évolutive. Les kinases ont ainsi été classées ce qui a permis d’établir un consensus objectif des groupes et des familles (Manning, 2002). La structure tertiaire peut aussi être utilisée dans ce contexte de classification et les bases de données CATH (Orengo et al., 1997) et SCOP2 (Andreeva et al., 2014) en sont des exemples.
Dans le contexte de développement d’un inhibiteur, étant donné que les protéines homologues à la cible primaire au niveau de la séquence ou de la structure tertaire sont susceptibles de lier des molécules semblables, des essais expérimentaux avec ces proches candidats doivent être menés afin d’optimiser la sélectivité de l’inhibiteur et prévenir la réactivité-croisée. Cependant, tel que mentionné précédemment, des protéines n’ayant aucune similarité de séquence ou de structure peuvent aussi montrer des profils de liaison similaires et donc l’optimisation de la sélectivité ne doit pas uniquement tenir compte des homologues au point de vue phylogénétique. Donc dans ce contexte, des méthodes capables de mesurer la similarité des facteurs déterminants de la fonction moléculaire sont plus garantes d’identifier des vrais positifs que les méthodes uniquement basées sur la séquence ou la structure.
Ces méthodes de similarités dites locales peuvent quand même être utiles lors de la comparaison des protéines ayant une haute similarité de séquence ou de structure, car ces protéines montrent parfois de petites différences au site de liaison ayant des effets drastiques sur la fonction, mais qui sont souvent silencieuses lors d’alignements de séquence ou de structure globale. Les applications des méthodes de similarités locales servent à la prédiction de fonction moléculaire, mais également dans un contexte de classification, d’identification de cibles croisées ou de cibles polypharmacologiques et pour la suggestion de remplacements bio-isostériques.
Méthodes de similarités de site de liaison
Une grande diversité de méthodes de détection de similarités locales et en particulier celles capables de comparer des sites de liaison a été développée, validée et appliquée dans différents contextes. Essentiellement, ces méthodes doivent d’abord créer une représentation réduite d’une protéine avec les éléments qui déterminent sa fonction moléculaire, tels la forme au site de liaison et l’arrangement tridimensionnel des acides aminés. Ensuite, elles doivent être capables de chercher des similarités entre les représentations de deux protéines et finalement de calculer un pointage des similarités. La façon la plus simple est d’utiliser les atomes du site de liaison et ceux-ci sont généralement réduits aux carbones alpha et/ou en type ou en groupes d’atomes afin d’optimiser les comparaisons. Les algorithmes de recherche et les fonctions de pointage des similarités sont adaptés au type de représentation et dépendent des informations incluses dans les modèles. La section qui suit résume de façon non exhaustive les méthodes de similarité publiées et vise à présenter la diversité des choix de conception.
Une méthode développée par le Chemical Computing Group et intégrée à PSILO (Feldman et Labute, 2010) utilise les carbones alpha répartis en classes selon l’identité du résidu et effectue une recherche exhaustive pour superposer tous les sous-ensembles de carbones alpha. Le pointage final est calculé en incluant les carbones beta et avec une fonction de pointage statistique normalisée avec une distribution de similarité entre 135 PDB aléatoires et 133800 sites de liaison. IsoCleft (Najmanovich et al., 2008), utilise tous les atomes du site de liaison et emploie l’algorithme d’appariement de graphe Bron & Kerbosh (Bron & Kerbosch, 1973) dont le temps computationnel augmente exponentiellement avec le nombre d’atome. Pour contrer cela, IsoCleft combine deux stratégies. Premièrement, il effectue une première recherche avec les carbones alpha et utilise lors d’une deuxième recherche tous les types d’atomes, mais uniquement ceux géométriquement proches des carbones alphas trouvés similaires à la première étape. Deuxièmement, l’algorithme de Bron & Kerbosch tend à trouver les plus grosses cliques en premier. Ici, une clique désigne une solution potentielle, soit un sous-ensemble d’atomes correspondants entre les deux protéines. Ainsi, d’arrêter à la première clique identifiée combinée à la recherche en deux
étapes permet un bon ratio performance/temps. IsoCleft utilise un pointage de Tanimoto qui considère la taille initiale des espaces de recherche. Sup-CK (Hoffmann et al., 2010) utilise les atomes convertis en types d’atomes et un algorithme du gradient ascendant cherche le meilleur chevauchement des densités d’atomes. Le pointage est calculé en utilisant notamment des informations additionnelles comme la charge partielle et le volume du site de liaison.
Soippa (Sequence Order Independant Protein Protein Alignment) utilise les carbones alpha issus d’une triangulation de Delaunay où chacun des carbones se voit attribuer un score de conservation calculé avec PSI-BLAST (Altschul et al., 1997). Chaque carbone alpha se voit attribuer un pointage d’un descripteur géométrique (Xie et Bourne, 2007), qui capture l’environnement géométrique local du carbone alpha et qui s’apparente en quelques sortes à une mesure d’enfouissement de l’atome. Soippa utilise l’appariement de graphe comme algorithme de recherche où chaque vertex du graphe associatif est une paire de carbones alpha, un de chaque protéine comparée, et qui ont une différence de descripteur géométrique d’au plus 50 unités. Chaque vertex se voit assigner un poids selon les profils de fréquence calculé avec PSI-BLAST. Les arêtes du graphe associatif relient les paires d’atomes si la différence des distances entre les atomes de la même protéine est moins de 2Å. Les solutions doivent avoir au moins trois paires d’atomes et le pointage est calculé selon la distance des profils avec une pénalité selon l’angle des vecteurs normaux et de la distance géométrique des atomes.
D’autres méthodes transforment les acides aminés en pseudocentres, c’est-à-dire en représentations fonctionnelles des acides aminés. Une méthode développée par (Wallach et Lilien, 2009) converti les acides-aminés d’un site de liaison en types donneur ou accepteur de pont hydrogène, en acide, en base, en centre aromatique et hydrophobe. Une optimisation de simplex Nelder-Mead est utilisée pour évaluer les transformations d’une protéine sur une autre et une mesure de chevauchement qui considère la forme et le type des centres superposés calcule le niveau de similarité. Ces pseudocentres sont placés à des
positions spécifiques des chaînes latérales. CavBase (Schmitt et al., 2002) assigne des pseudocentres donneur, accepteurs, donneur-accepteur, PI (aromatique) ou aliphatique aux acides-aminés. Également, un ensemble de points décrit la surface de la cavité et chaque point se voit assigner le type du pseudocentre le plus proche selon des contraintes de distance et d’angle (pour les interactions de donneurs et d’accepteurs). Un graphe associatif est construit où chaque vertex correspond à une paire de pseudocentres compatibles. Les arêtes relient les vertices du graphe si les pseudocentres de la même cavité parmi les deux paires sont à moins de 12Å l’un de l’autre et si la différence des distances est moindre que 2Å. Une particularité de CavBase est que la fonction de pointage dépend de l’importance du chevauchement de points de surface du même type et non simplement du nombre de pseudocentres trouvés. Les 100 premières cliques les plus larges selon le nombre de pseudocentres sont repointées avec le chevauchement de surface. ProBis (Konc et Janežič, 2010) est une méthode très similaire à CavBase, mais qui décompose le site de liaison de chaque protéine en n sous graphe, où n représente le nombre de vertex (pseudocentres) de la protéine. Chaque sous-graphe a donc un pseudocentre de référence et une matrice de distance des vertices à moins de 15Å de la référence est construite. Lorsqu’une protéine est comparée à une autre, toutes les paires de sous-graphes sont comparées via leurs matrices de distance et la similarité calculée par la méthode décrite dans (Konc et Janežič, 2007) permet de filtrer les paires de sous-graphes qui seront ensuite analysés par l’algorithme de Bron & Kerbosch. Cette étape de filtrage est un autre exemple qui permet d’obtenir un ratio performance/temps de calcul appréciable.
Plusieurs méthodes présentées jusqu’à présent utilisent les algorithmes d’appariement de graphe qui sont très gourmands en temps computationnel. RAPMAD (Krotzky et al., 2014a) est une modification importante de CavBase qui démontre que l’appariement de graphe peut être remplacé par un algorithme plus rapide. Cependant, une des différences avec CavBase est que le pseudocentre aromatique est décomposé en deux nouveaux pseudocentres, un centre de cycle aromatique et un centre d’électrons π et il y a l’ajout d’un pseudocentre ion métallique donnant un total de 7 types. Le centre de masse d’un type constitue le premier point de référence et le pseudocentre le plus près du centre de masse et
du même type constitue le deuxième point de référence. Ainsi, il y a un total de 14 points de référence. Les distributions des distances euclidiennes entre chaque point de référence et les pseudocentres du même type sont calculées et normalisées à l’aide de la fréquence d’observation de chaque pseudocentre. Par exemple, les pseudocentres d’ions métalliques sont observés moins fréquemment (0.1%) et ont donc un poids plus important dans le pointage. Les distances euclidiennes sont groupées en intervalles de 0.4Å. Le pointage entre deux protéines consiste à calculer la divergence de Jensen-Shannon entre les deux ensembles de 14 histogrammes mis un derrière l’autre. RAPMAD n’est pas le seul algorithme qui encode les propriétés du site de liaison dans des distributions de distance. PocketMatch (Yeturu et Chandra, 2008) utilise une méthode similaire avec un ensemble de 90 listes de distances. Tous les atomes des chaînes latérales sont divisés en 5 groupes : A,V,I,L,G,P - K,R,H - D,E,Q,N - Y,F,W et C,S,T. Les chaînes latérales sont réduites en seulement trois points de référence: les carbone alpha, beta et un centre de masse de tous les atomes. Si chacun des trois points peut être un des cinq groupes, il y a donc 90 combinaisons possibles de paires. Chaque distance euclidienne entre tous les points est calculée et placée dans une des 90 combinaisons et cette liste de distance est mise en ordre ascendante facilitant l’alignement et le pointage qui se basent sur un algorithme gourmand qui tient compte de la cardinalité de chaque groupe et du nombre total de distances équivalentes et de la fréquence de chaque groupe.
SuMo (Jambon et al., 2003) réduit les acides aminés d’un site de liaison à un ensemble de points qui représentent différents groupes stéréochimiques (guanidium, amide, acyl, thiol, glycine, aromatique, ammonium, etc.) et à chaque point une densité atomique est calculé qui est une mesure indirecte du niveau d’enfouissement. Aucun groupe stéréochimique hydrophobe n’est utilisé, mais les auteurs mentionnent que la mesure de densité compense pour cela. Une particularité de la méthode est qu’au lieu de construire des graphes de groupes stéréochimiques pour l’étape de la recherche, les graphes sont construits avec des triplets de groupes. Ainsi ce sont des paires de triplets de groupes et non des paires d’atomes qui sont comparées ce qui diminue significativement le temps computationnel
d’une comparaison. La fonction de pointage intègre la distance euclidienne des paires de triangles d’une solution et une pénalité selon la différence de densité des atomes.
eMatchSite (Brylinski, 2014) utilise un algorithme d’apprentissage automatique pour prédire les distances entre chaque paire de carbone alpha de deux protéines à l’aide d’un modèle à 7 descripteurs assignés à chaque carbone alpha : le profil de séquence via PSI-BLAST (Altschul et al., 1997), le profil de structure secondaire PSIPRED (Jones, 1999), un pointage d’hydrophobicité calculé selon 20 méthodes différentes, une probabilité de liaison calculée avec eFindSite (Brylinski et Feinstein, 2013), la distribution spatiale de résidus adjacents, l’entropie de séquence, c’est-à-dire la variabilité des acides aminés à une position selon PSI-BLAST et une similarité chimique d’atomes de ligands prédits adjacents aux carbones alpha avec eFindSite. L’alignement optimal des carbones alpha de deux sites de liaison et le score final est calculé à l’aide de l’algorithme de Kuhn-Munkres qui garantit de trouver l’ensemble unique de paires d’atomes qui produit la plus petite distance totale parmi toutes les combinaisons. eMatchSite est le seul algorithme qui utilise l’apprentissage automatique, qui est entraîné sur l’ensemble de données publié par Soippa.
SiteEngine (Shulman-Peleg et al., 2004) assigne des pseudocentres à chaque acide aminé de façon similaire à CavBase, mais avec des centres chargés positivement et négativement respectivement pour les résidus Lys, Arg et His ainsi que pour Asp et Glu et qui sont utilisés seulement lors de l’étape de pointage. Une surface de conolly est générée et pour chaque point de cette surface un type de pseudocentre est assigné et un indice de courbure de surface est calculé. SiteEngine utilise un algorithme de hachage géométrique ou chaque triplet de pseudocentres, qui a des longueurs d’arête plus petite qu’un certain seuil, est enregistré dans une table de hachage avec la courbure. Lorsque deux protéines sont comparées, les matrices de transformation qui superposent chaque paire de triplets, dont les pseudocentres sont du même type et avec des courbures équivalentes sont utilisées lors de la recherchées et pour pointer les superpositions des surfaces sur lesquelles sont projetés l’information des pseudocentres. Une autre méthode fonctionne de façon similaire,
c’est-à-dire avec des tables de hachage, mais en utilisant l’identité des atomes au lieu de pseudocentres et la fonction de pointage ne considère aucune surface, mais seulement le chevauchement des atomes (Brakoulias et Jackson, 2004).
KRIPO (Wood et al., 2012) place des centres fonctionnels (donneur, accepteur, aromatique pi, hydrophobe ainsi que des charges positives et négatives) à une certaine distance des atomes fonctionnels et dans une direction optimale pour les ponts hydrogènes, par exemple le long de l’axe C=O d’un carbonyl. La représentation consiste à lister les combinaisons de 2 et 3 centres qui ont été brouillés. En d’autres mots, la même combinaison est ajoutée à la liste plusieurs fois en variant la distance relative des centres. C’est la façon qu’ont trouvée les auteurs de la méthode pour intégrer une variabilité géométrique. Afin de faciliter la recherche, les distances entre les centres de groupes ont été normalisées en intervalles réguliers. Le pointage utilise une version modifiée du coefficient de Tanimoto, celle proposée par Fligner et Verducci (Fligner et al., 2002) qui tient compte de la densité moyenne des groupes de centres par site de liaison afin de réduire le biais lié à la différence de taille des sites comparés.
Comme décrit plus haut, la plupart des méthodes intègrent des informations en plus du type d’atome et de leur position, comme la courbure de la surface adjacente, des profils de conservation position-spécifique ou des mesures d’angle pour les interactions directionnelles. eMatchSite, présenté ci-haut, utilise sept descripteurs pour chaque carbone alpha. PocketFEATURE (Liu et Altman, 2011) utilise le système FEATURE (Wei et al., 1997) contenant 80 descripteurs physico-chimiques dont l’identité du résidu, charge, structure secondaire, volume de van der Waals, hydrophobicité, facteur-b, accessibilité au solvant. Aux atomes fonctionnels des résidus d’un site de liaison, la présence des 80 descripteurs est évaluée dans 6 rayons concentriques à l’atome de référence pour un rayon total de 7.5 Å formant un vecteur de 480 propriétés appelé microenvironnement. Lors de la comparaison de deux cavités, la similarité de toutes les paires de microenvironnements est évaluée à l’aide d’un coefficient de Tanimoto et deux microenvironnement sont similaires
si leur coefficient de Tanimoto est à moins d’une déviation standard l’un de l’autre. Un total de 22008 microenvironnements d’un ensemble de 1160 sites de liaisons d’un ensemble non redondant de la PDB est utilisé pour calculer une distribution de fond afin de dériver une similarité moyenne et une déviation standard. Pour accélérer les comparaisons, seuls les microenvironnements centrés sur des acides aminés du même groupe (chargés positivement, négativement, polaires, non-polaires et aromatique) sont considérés. Le score final est la somme des paires de microenvironnement les plus similaires et est normalisé avec la distribution de fond. Une caractéristique unique de cette méthode est qu’aucune contrainte géométrique n’est appliquée lors de la recherche. La composante géométrique est capturée dans les rayons concentriques des microenvironnements et les auteurs suggèrent que cette particularité leur permet d’être plus robustes face à des cas, comme avec le FAD, ou le même ligand montre une très grande flexibilité géométrique entre deux modes de liaison. Une méthode publiée en 2010 (Milletti et Vulpetti, 2010) utilise aussi un système de sphères concentriques, mais avec les types d’atomes comme unique descripteur. Les auteurs n’ont pas rapporté la robustesse de la méthode face à des modes de liaison à haute flexibilité géométrique.
La méthode eF-Site (Kinoshita et Nakamura, 2003) utilise une surface de conolly où à chaque point de la surface, les équations de poisson boltzmann sont utilisées pour calculer un potentiel électrostatique. À chaque point, la courbure et un indice hydrophobe sont également calculés avec l’échelle de Kyte et Doolittle (Kyte et Doolittle, 1982). L’algorithme d’appariement de graphe Bron & Kerbosch est utilisé pour la recherche avec un critère de différence pour construire les arêtes de 1.5Å. SiteAlign est une méthode originale: la représentation consiste au placement d’un seul polyèdre à 80 faces au centre de masse des atomes du site de liaison. Huit descripteurs sont projetés à partir des carbones beta sur les faces du polyèdre: la distance entre la face et le carbone beta, l’orientation de la chaîne latérale c’est-à-dire si la chaîne latérale pointe vers l’intérieur ou l’extérieur du polyèdre, la taille de la chaîne latérale, si le résidu du carbone beta participe comme donneur ou accepteur de pont hydrogène, la charge de l’atome ainsi que le caractère hydrophobe et aromatique. L’algorithme de recherche consiste à appliquer des translations
et des rotations d’une sphère dans sa cavité, de recalculer les descripteurs puis de comparer la similarité des faces avec la sphère de la deuxième protéine qui elle reste fixe. Le pointage est la somme normalisée des différences des descripteurs des triangles du polyèdre.
GRID-FLAP (Fingerprints for Ligands And Proteins) (Baroni et al., 2007) est une méthode de détection de similarités qui utilise GRID (Goodford, 1985). GRID est une méthode computationnelle pour identifier les régions favorables énergétiquement (appelés MINI) pour 5 sondes : donneur et accepteur de ponts hydrogène, charge positive, charge négative et DRY la sonde hydrophobe. FLAP utilise une fonction pour garder le sous-ensemble de MINIs qui représente le mieux leur dispersion dans l’espace. FLAP utilise toutes les combinaisons de 4 MINIs d’une cavité, formant des tétraèdres, comme base de comparaison. Chaque tétraèdre est encodé dans un vecteur à une dimension contenant les 6 distances du tétraèdre, les types de sondes avec leurs coordonnées et la somme des énergies de chaque sonde. Chaque paire de tétraèdres entre deux sites de liaisons qui ont des sondes correspondantes et une différence de distance maximale de 1Å pour leurs 6 arêtes est utilisée pour superposer les deux MIF. Suite à la superposition, la similarité est calculée suite au chevauchement des sondes.
Problématique et hypothèses Représenter la fonction moléculaire
Les méthodes de détection de similarité utilisent un continuum d’éléments pour représenter les sites de liaison, allant de la séquence (p. ex. avec les données de PSI-BLAST) à la structure (p. ex. avec les descripteurs géométriques) et en passant par les atomes de carbones alpha du squelette peptidique, puis par les atomes des chaînes latérales, à leurs groupements fonctionnels seulement, jusqu’à la représentation de la surface électrostatique. Les méthodes cherchent ensuite à détecter les similarités parmi ces éléments entre deux sites qui peuvent ensuite être interprétés en une similarité de fonction moléculaire. Une fonction moléculaire est essentiellement définie par la présence d’atomes (ou de types
d’atomes) à des positions précises du volume d’une cavité. Ainsi, de représenter les sites de liaison par une distribution potentielle des atomes qui peuvent se retrouver dans le volume de leur cavité représenterait plus directement la fonction moléculaire et serait susceptible d’améliorer la performance des méthodes de détection de similarité.
Le côté d’où provient une chaîne latérale vers un groupe d’atome du ligand n’est pas toujours important. Une lysine peut provenir de deux directions opposées, relativement à une molécule d’ATP dans deux sites de liaison différents, mais au final, l’énergie potentielle calculée à la position d’un groupement phosphate de l’ATP sera sensiblement le même. La position des amines de ces deux lysines sera différente, si l’une provient de la gauche et l’autre lysine de la droite et cette différence de position est d’autant plus importante pour les carbones beta et alpha, qui sont plus loin du ligand. Le principe s’applique également pour les interactions aromatiques d’empilement, mais cela est discutable pour les ponts hydrogènes, qui sont des interactions directionnelles. Cependant, même la direction relative au ligand du donneur ou de l’accepteur de la protéine n’est pas tenue d’être identique, car, selon la flexibilité du ligand, des chaînes latérales et de la chaîne principale, celui-ci pourrait se réorienter légèrement pour accommoder ces différences de directionalité. Les représentations qui se basent sur les positions des atomes du site de liaison sont donc susceptibles d’échouer à identifier certaines similarités. Donc de mesurer l’effet combiné des atomes des sites de liaison dans le volume de la cavité engendre une représentation plus proche de la fonction moléculaire, ce qui est susceptible d’améliorer la performance des méthodes de détection de similarité.
Champs d’interaction moléculaire
Un champ d’interactions moléculaires ou MIF pour Molecular Interaction Field décrit la distribution des potentiels énergétiques de différents groupes chimiques et est donc susceptible d’être la méthode idéale pour représenter la fonction moléculaire dans le volume d’une cavité. Présentement, malgré les dizaines de méthodes différentes, il n’y a que la méthode GRID-FLAP qui utilise une représentation de MIF. Les MIFs sont
généralement calculés à l’aide d’équations qui tiennent compte du potentiel de coulomb et du potentiel de Lennard-Jones. Dans GRID-FLAP le potentiel comprend un terme pour les interactions électrostatiques, de pont hydrogène, Lennard-Jones et pour les contributions entropiques. En général, les MIFs sont utilisés pour l’optimisation d’inhibiteurs lors d’analyses de 3D-QSAR (Three-Dimensional Quantitative Structure Activity Relationship) comme les analyses comparatives de champs moléculaires CoMFA (Comparative
Molecular Field Analysis). Elles permettent de corréler les changements d’affinité ou
d’activité biologique avec des changements structuraux et ainsi connaître les groupements chimiques de la molécule qui influent sur la sélectivité et la spécificité (da Silva et al., 2004, Kastenholz et al., 2000). Ces méthodes consistent d’abord à superposer les petites molécules pour lesquelles les données d’affinité ou d’activité sont connues. Pour plus de précision, la superposition doit se baser sur les conformations observées dans les structures cristallines ou RMN des molécules complexées avec la cible d’intérêt. Le potentiel de chaque sonde est calculé à chaque intersection d’une grille remplissant le volume des molécules superposées, formant une matrice où chaque rangée représente une des petites molécules et chaque colonne une combinaison position-sonde. Souvent, la méthode des moindres carrés est utilisée pour réduire la matrice à un modèle mathématique qui décrit les variations d’affinité et d’activité en fonction des potentiels mesurés avec les différentes sondes. On peut aussi appliquer les méthodes des moindres carrés partiels ou PLS (Partial
Least Square) ou la méthode GOLPE (Generating Optimal Linear PLS Estimation) pour
identifier seulement les variables les plus descriptives du modèle avant d’appliquer la méthode des moindres carrés (Cruciani et Watson, 1994). Les méthodes de 3D-QSAR permettent d’obtenir des résultats faciles à interpréter visuellement pour optimiser les groupements chimiques des inhibiteurs.
Objectifs
GRID-FLAP est la seule méthode, qui utilise les champs d’interaction moléculaire. Cette méthode a récemment été intégrée à BioGPS (Siragusa et al., 2015), un algorithme qui prépare les structures, détecte les cavités, mesure les MIFs et effectue les comparaisons. Il est possible que certains choix de conception de cette méthode limitent les performances.
Tel que mentionné par les auteurs, chaque site de liaison peut comporter des millions de quadruplets et donc des milliards des paires de quadruplets sont générés lors d’une comparaison. Un seuil de 1Å a donc été utilisé, tel que mentionné ci-haut, pour optimiser le ratio performance/temps (Siragusa et al., 2015) ce qui est relativement contraignant considérant la flexibilité intrinsèque des structures de protéine et des ligands. Sa performance a été évaluée lors d’une classification de 23 kinases avec une analyse des composantes principales (Sciabola et al., 2010), d’enzymes selon l’identificateur EC et sur la capacité à retrouver, parmi un ensemble de sites, celui de la protéine SERCA (Sarcoplasmic reticulum Ca2+ ion channel ATPase) en utilisant un site similaire soit celui du récepteur à l’estrogène alpha (Siragusa et al., 2015).
La performance d’une méthode basée sur les MIFs, comme GRID-FLAP, n’a donc jamais été comparée à celles de méthodes ayant une représentation dite classique (i.e. qui utilisent des atomes ou des pseudocentres) avec des bancs d’essai. GRID-FLAP étant la propriété de Molecular Discovery, sa disponibilité est limitée et il est donc difficile d’effectuer de telles analyses avec ce logiciel. De plus, l’impact des choix de conception, notamment ceux liés à la détection des cavités, à la définition des sites de liaison, ou des paramètres de tolérance à la variabilité géométrique, est susceptible d’être différent pour les méthodes basées sur les MIFs que pour les méthodes avec des représentations dites classiques. Il est donc nécessaire de développer une nouvelle méthode de détection de similarités basée sur les MIFs et d’évaluer ses performances relativement à d’autres méthodes et d’analyser les limites d’une telle méthode. De plus, il serait intéressant de rendre la méthode disponible gratuitement à la communauté scientifique.
Objectif #1 – Développement d’une nouvelle méthode (Chapitre 2, article 1)
Le premier objectif est de développer une méthode de détection de similarité de sites de liaison qui utilise les MIFs comme représentation. La méthode, baptisée IsoMif suivant une nomenclature similaire à son logiciel prédécesseur IsoCleft, utilisera un potentiel exponentiel simplifié ainsi qu’une matrice d’interaction à 6 sondes : donneur et accepteur de pont hydrogène, charge positive et négative, aromatique et hydrophobe. Certaines modifications à l’algorithme d’appariement de graphe Bron & Kerbosch ont été utilisées
dans IsoCleft pour réduire le compromis performance/temps. Ainsi il a été possible de conserver une certaine précision avec un temps de calcul permettant d’effectuer des analyses à haut débit. Plus précisément, une grille à résolution variable permettra de moduler la taille du graphe associatif et la tendance qu’a l’algorithme de Bron & Kerbosch à identifier les plus grosses cliques en premier sera exploitée pour optimiser le ratio performance/temps de l’étape de recherche. L’information des vertices trouvés similaires sera utilisée pour calculer un score de similarité basé sur une mesure de Tanimoto. Nous porterons une attention particulière à l’étape de visualisation des similarités qui facilite l’interprétation des résultats.
Objectif #2 – Validation et comparaison des performances (Chapitre 2, article 1)
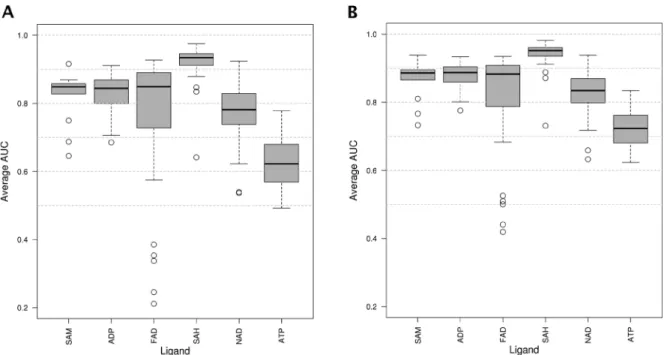
IsoMIF sera validé sur des jeux de données utilisés pour évaluer les performances d’autres méthodes classiques et des jeux de données additionnels seront utilisés pour mesurer la performance comme l’ensemble scPDB et PDBbind, deux jeux de données de grande taille contenant des complexes protéine-ligands. La similarité de séquence sera utilisée comme méthode contrôle. L’évaluation de la performance se fera à l’aide de courbes ROC et l’aire sous la courbe (AUC) principalement parce que c’est la mesure utilisée par la majorité des auteurs et donc nécessaire pour comparer les performances. Nous pourrons calculer d’autres mesures de performance comme les facteurs d’enrichissement.
La définition du site de liaison est un aspect important lors de l’évaluation de la performance des méthodes. Elle dicte le niveau d’information superflue ajouté et qui peut brouiller la performance. Ce n’est pas un aspect fréquemment discuté ou validé dans la littérature. Nous évaluerons la performance selon cette variable afin que les données soient disponibles à des fins de comparaison ultérieurement. Nous évaluerons l’impact d’autres paramètres sur la performance d’IsoMif, notamment en lien avec la flexibilité de la protéine.
Objectif #3 - Développement d’une interface en ligne (Chapitre 3, article 2)
Nous développerons une interface en ligne afin de rendre l’outil disponible à la communauté scientifique. Plus précisément nous voulons permettre à un utilisateur, qui ne possède pas de connaissances en informatique, de comparer des cavités de sa protéine à plusieurs jeux de donnée de champ d’interaction moléculaire précalculés ou à ses propres cavités. L’interface doit permettre de raffiner les cavités directement dans le navigateur sans avoir besoin d’utiliser un logiciel externe, car cela est parfois limitant pour les utilisateurs non experts. Les résultats seront montrés directement en ligne dans un tableau avec des images montrant les similarités. De plus, les résultats doivent être disponibles sous forme de session PyMOL afin de permettre aux utilisateurs de visualiser en détail les similarités.
Objectif #4 – Application à large échelle (Chapitre 4)
Nous effectuerons une analyse à grande échelle de cibles croisées pour un ensemble de ligands en comparant leur site de liaison à un ensemble de sites de liaison potentiels. Des simulations d’arrimage moléculaire des ligands dans les cibles prédites complémenteront les cibles croisées les plus similaires trouvées avec IsoMif. L’objectif de cette analyse et de générer des hypothèses qui pourront être repris par d’autres dans deux contextes: le repositionnement de médicaments existants pour de nouvelles utilisations et l’explication d’interactions secondaires pouvant expliquer les effets secondaires observés.
C
HAPITRE2
-
A
RTICLE1
Detection of Binding Site Molecular Interaction Field Similarities Auteurs de l’article: Chartier, M. et Najmanovich, R.
Statut de l’article: Publié. Référence : Chartier, M., & Najmanovich, R. (2015). Detection
of Binding Site Molecular Interaction Field Similarities. Journal of Chemical Information
and Modeling, 150717083947004. http://doi.org/10.1021/acs.jcim.5b00333
Avant-propos: J’ai effectué le développement de la méthode, la préparation et l’analyse
des données sous la supervision de Rafael Najmanovich. J’ai rédigé l’article au complet sous la supervision de Rafael Najmanovich.
Résumé : Les méthodes de détection de similarités de sites de liaison sont utilisées pour
détecter des conditions physico-chimiques menant à une même fonction moléculaire. Bien que les champs d’interaction moléculaire représentent plus directement la fonction moléculaire, la majorité des méthodes existantes utilisent des représentations basées sur les atomes.
Nous avons donc développé IsoMif, une méthode basée sur les champs d’interaction moléculaire. IsoMif utilise un potentiel simplifié, 6 sondes représentant les interactions intermoléculaires aromatiques, donneur et accepteur de pont hydrogène, à charge positive et négative ainsi que le potentiel hydrophobe. L’algorithme d’appariement de graphe de Bron & Kerbosch est utilisé pour identifier les similarités et le pointage se base sur le coefficient de Tanimoto.
La méthode a été validée à l’aide de 6 jeux de données et la performance a été comparée à celle d’autres méthodes. IsoMif performe mieux en moyenne et de façon plus robuste. L’effet de la définition du site de liaison ainsi que d’autres paramètres et l’introduction de la flexibilité ont été évalués. Deux exemples d’applications sont donnés.
Abstract
Protein binding-site similarity detection methods can be used to predict protein function and understand molecular recognition, as a tool in drug design for drug repurposing and polypharmacology, and for the prediction of the molecular determinants of drug toxicity. Here, we present IsoMIF, a method able to identify binding site molecular interaction field similarities across protein families. IsoMIF utilizes six chemical probes and the detection of subgraph isomorphisms to identify geometrically and chemically equivalent sections of protein cavity pairs. The method is validated using six distinct data sets, four of those previously used in the validation of other methods. The mean area under the receiver operator curve (AUC) obtained across data sets for IsoMIF is higher than those of other methods. Furthermore, while IsoMIF obtains consistently high AUC values across data sets, other methods perform more erratically across data sets. IsoMIF can be used to predict function from structure, to detect potential cross-reactivity or polypharmacology targets, and to help suggest bioisosteric replacements to known binding molecules. Given that IsoMIF detects spatial patterns of molecular interaction field similarities, its predictions are directly related to pharmacophores and may be readily translated into modeling decisions in structure-based drug design. IsoMIF may in principle detect similar binding sites with distinct amino acid arrangements that lead to equivalent interactions within the cavity. The source code to calculate and visualize MIFs and MIF similarities are freely available.
Introduction
The identification of similarities between proteins has many practical applications. Sequence similarity algorithms like BLAST (Mount, 2007) allow one to quickly retrieve homologous proteins and transfer potential functional annotations from the target to the query protein. Sequence similarity also enables the construction of phylogenetic trees grouping proteins into families, for example with kinases (Manning, 2002), to view functional information within an evolutionary context (Chartier et al., 2013). Whereas traditionally the function of a protein was well-characterized biochemically prior to the elucidation of its structure, with the advent of structural genomics projects in the past decade, there has been an influx of proteins for which the structure is known but not their function. Because sequence is less conserved than structure (Rost, 1999), comparing two proteins using sequence alone ignores structural information that is relevant for function. A number of well-established methods such as DALI (Holm et Sander, 1997) measure global structural similarities, which coupled with databases like SCOP (Andreeva et al., 2014) or CATH (Sillitoe et al., 2013) can be used to understand the cellular (biological processes) or molecular (molecular interactions) functions of a protein. Finally, meta-servers such as ProFunc (Laskowski et al., 2005) combine a number of sequence- and structure-based methods to predict protein function from structure.
Even when the function of a protein is known, the detection of similarities has important applications. From a drug design perspective, detecting proteins that are similar to the drug target is important to prevent cross-reactivity and the potential associated side effects. In such cases, sequence and structural similarities can fail for two reasons. First, divergent evolution may introduce mutations deemed minor at the level of sequence or structure, but with drastic effects locally in the binding site, altering the molecular function, e.g., shifting substrate specificity. Second, convergent evolution can bring proteins unrelated by sequence or structure to acquire the same molecular function. In these two cases, the similarities act at a more local scale and affect the physicochemical environment. Therefore, a solution is to use methods that can detect local physicochemical similarities in binding pockets.
Such methods can help predict molecular function, identify cross-reactivity or polypharmacological targets, predict binding fragments, and repurpose existing drugs. For example, IsoCleft (Najmanovich et al., 2008, Kurbatova et al., 2013) is graph-matching-based method for the detection of 3D atomic binding site similarities that was used to predict function for structural genomics proteins (Han et al., 2010, Bakolitsa et al., 2010), reclassify members of the human cytosolic sulfotransferase family (Allali-Hassani et al., 2007, Najmanovich et al., 2000), and analyze the druggability of histone methyltransferases (Campagna-Slater et al., 2011) based on binding site similarities. SOIPPA (Xie et Bourne, 2008a) predicted off-targets for CETP inhibitors in agreement with experimental assays, and the authors explained adverse side effects observed in clinical trials using a systems biology approach (Xie et al., 2009). FragFEATURE (Tang et Altman, 2014) uses the FEATURE microenvironments (Halperin et al., 2008) combined with a knowledge-based approach to predict binding fragments for a target protein. CavBase detected similarities between COX-2 and carbonic anhydrase protein structures both known to bind COX-2 selective Celecoxib inhibitor (Weber et al., 2004). The algorithms behind these and the numerous other methods for the prediction of binding site similarities can be divided into three components: representation, search, and scoring.
With respect to representations, many methods transform the protein into a simplified representation and sometimes incorporate additional information. Mostly, proteins are represented using Cα atoms, functional atoms, pseudocenters, or electrostatic surfaces, but some use all atoms. For example, SOIPPA ( Xie et Bourne, 2008b) and Psilo (Feldman et Labute, 2010) use Cα atoms, IsoCleft (Najmanovich et al., 2008, Kurbatova et al., 2013) uses a two-step Cα/all-atom process, CavBase (Schmitt et al., 2002) places pseudocenters in the vicinity of amino acids using distance and angle cutoffs, SiteEngine (Shulman-Peleg et al., 2004) proceeds similarly but without angle considerations for hydrogen bonds and eF-site (Kinoshita et Nakamura, 2003) uses a Connolly surface where the electrostatic potential is calculated. Other more unique forms of representation exist; pocketFEATURE (Liu et Altman, 2011) transforms important binding site atoms into micro environments that consist of physicochemical descriptors on concentric spherical shells, siteAlign (Schalon et al., 2008) uses an 80-face polyhedron onto which descriptors are projected and