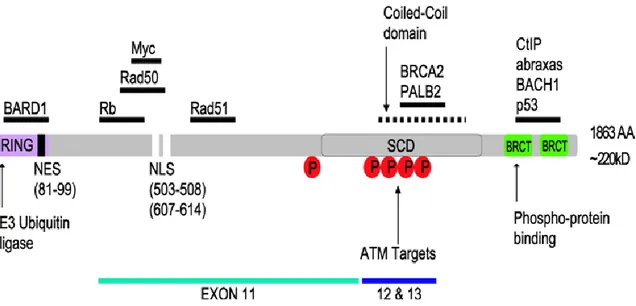
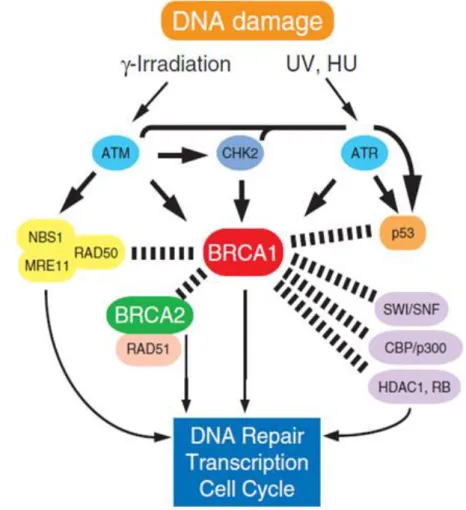
BRCA1 et réponse aux agents anticancéreux dans les hépatocarcinomes
122
0
0
Texte intégral
Figure
+7
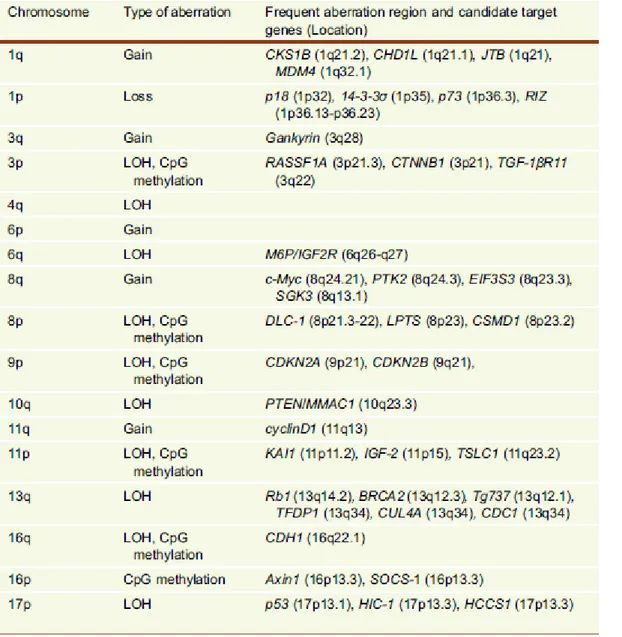
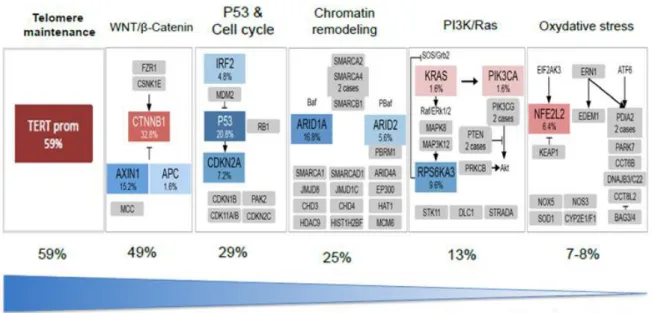
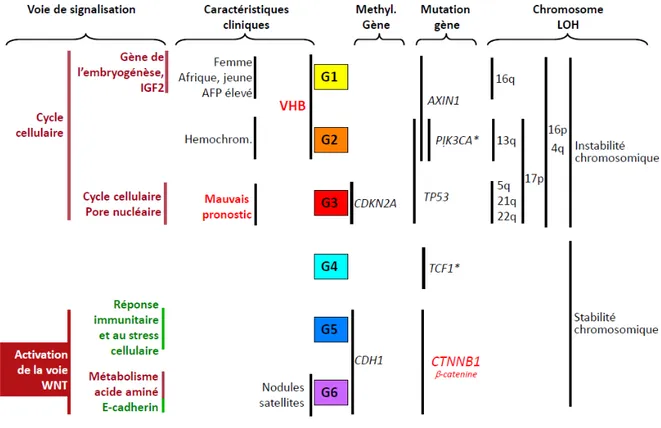
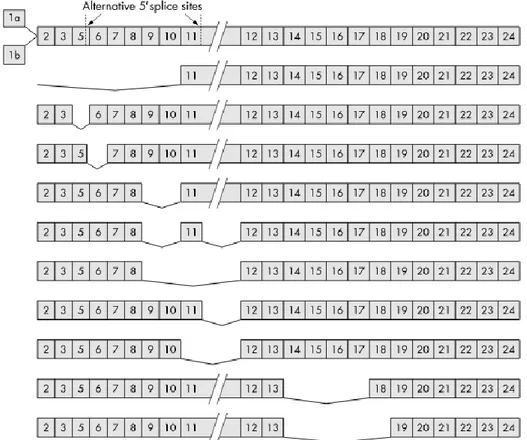
Documents relatifs
