Putative Implication of
␣-Amylase Loop 7 in the
Mechanism of Substrate
Binding and Reaction
Products Release
V. Tran 1 Institut Pasteur, Unite´ de Biochimie Structurale, 25 rue du Dr Roux, 75724 Paris Cedex 15, France 2 Unite´ de Biotechnologie, Biocatalyse et Biore´gulation, Faculte´ des Sciences et techniques 2, rue de la Houssinie`re, BP 92208, 44322 Nantes Cedex 03, France Received 2 April 2002; accepted 5 April 2004Published online 21 July 2004 in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/bip.20096
Abstract: ␣-Amylases are widespread endo-enzymes involved in the hydrolysis of internal ␣-(1,4)
glycosidic linkages of starch polymers. Molecular modeling of amylose–amylase interactions is a step toward enzymatic mechanism understanding and rational design of new enzymes. From the crystallographic complex of barley ␣-amylase AMY2-acarbose, the static aspects of amylose– amylase docking have been characterized with a model of maltododecaose (DP12) (G. Andre´, A. Bule´on, R. Haser, and V. Tran, Biopolymers 1999, Vol. 50, pp. 751–762; G. Andre´ and V. Tran, Special Publication no. 246 1999, The Royal Society of Chemistry, H. J. Gilbert, G. J. Davies, B. Henrissat, and B. Svensson, Eds., Cambridge, pp. 165–174). These studies, consistent with the experimental subsite mapping (K. Bak-Jensen, G. Andre´, V. Tran, and B. Svensson, Journal of
Biological Chemistry, to be published), propose a propagation scheme for an amylose chain in the
active cleft of AMY2. The topographical overview of␣-amylases identified loop 7 as a conserved segment flanking the active site. Since some crystallographic experiments suspected its high flexibility, its putative motion was explored through a robotic scheme, an alternate route to dynamics simulations that consume CPU time. The present article describes the characteristics of the flexibility of loop 7: location and motion in AMY2. A back-and-forth motion with a large amplitude of more than 0.6 nm was evaluated. This movement could be triggered by two hinge residues. It results in the loop flipping over the active site to enhance the docking of the native helical substrate through specific interactions, it positions the catalytic residues, it distorts the substrate towards its transition state geometry, and finally monitors the release of the products after hydrolysis. The residues involved in the process are now rational mutation points in the hands of molecular biologists. © 2004 Wiley Periodicals, Inc. Biopolymers 75: 95–108, 2004
Correspondence to: G. Andre´; email: gandre@pasteur.fr
Biopolymers, Vol. 75, 95–108 (2004) © 2004 Wiley Periodicals, Inc.
Keywords: molecular modeling; barley ␣-amylase; loop motion; substrate distortion; robotic
calculations; hinge residues
INTRODUCTION
Most proteins display a multiple domain folding. Large and relative motions of domains are often re-sponsible for protein flexibility, and more generally, for protein function. Indeed, domain motions have been shown as essential for a variety of protein func-tions including catalysis, regulation of activity, trans-port of metabolites, formation of protein assemblies, and cellular locomotion.1–3 Domains often close around the binding site through ligand-induced con-formational transition.4Generally, the binding of sub-strates stabilizes a protein structure in its “closed” conformation, and conversely, the absence of sub-strate favors the protein structure in its “open” state.1 Until now, most experimental information on the mechanism of domain motions has come from x-ray diffraction that resolved opened and/or closed domain structures of proteins. The role of a domain closure during catalysis is often to discard useless water mol-ecules, to position the catalytic residues in their at-tacking geometry,5 and to distort the substrate to-wards a transition state conformation.1,4
Gerstein et al. have studied1the partial mobility of different domains in proteins. From a survey of all the experimentally observed domain movements in pro-teins, they have defined two low-energy conforma-tional changes: hinge and shear motions. Particularly, among the studied domain movements, hinge motion has been clearly shown for the triose phosphate isomerase6(TIM), the tryptophan synthase ␣22,4,7 and the inosine 5⬘-monophosphate dehydrogenase8 where these enzymes show a typical⍀ loop,9,10 con-necting one-sheet to an ␣-helix of the typical TIM barrel folding. In the first two cases, a weak electron density in the region of loop 6 (because located be-tween strand 6 and helix 6 of the ␣/ barrel) com-bined with fairly high temperature factors, when the proteins are in their native state, implies a high degree of conformational mobility for this loop. In the latter case, loop 6 is clearly visible in the electron density map and is in a different conformation from those in the substrate and substrate analogue-bound structures. It is worth mentioning that⍀ loops are known to be often involved in protein function, folding, and sta-bility, as well as in molecular recognition.10 In the case of the TIM enzyme, loop 6 has been suspected to move as a rigid lid thanks to two hinges. Its motion helps to cover the active site and to reinforce the binding of the substrate.2,6In the tryptophan synthase
␣22 enzyme, closure of loop 6 isolates the active site of the␣-subunit from solvent and results in interac-tion between specific and identified residues.4,7 In
both cases, site-directed mutations confirmed the as-sumptions and identified residues of the loop as es-sential for the catalytic activity. Finally, in the inosine 5⬘-monophosphate dehydrogenase8 case, it has been shown that loop 6 follows a similar pattern of hinge-body motion and indicates that the protein may be using this loop to bind and sequester substrate and to recruit an essential catalytic residue.
The main interesting and connecting point with TIM isomerase, tryptophan synthase␣22, and ino-sine 5⬘-monophosphate dehydrogenase is that ␣-amy-lases exhibits the same (␣/)8barrel topology with a
similar loop despite completely different enzymatic functions. X-ray diffraction studies have been per-formed on the wide area of␣-amylase origins such as
Aspergillus oryzae,11,12 Aspergillus niger,13Bacillus licheniformis,14 Bacillus subtilis,15Alteromona halo-planctis,16yellow meal worm,17porcine18,19and hu-man pancreas,20 human saliva,21,22 and barley.23,24
Their native or complex state have revealed experi-mental evidence for a highly conserved and flexible loop flanking the active site. The loop is suspected to flip over the active site toward a “closed” thus inac-cessible position. In the case of PPA, porcine pancre-atic␣-amylase, this loop has been seen in two posi-tions, “open” and “closed” in the crystal, depending respectively on the presence or absence of the com-petitive inhibitor acarbose. In particular, a significant displacement of 0.5 nm has been measured for
HPPA305 of the loop.18,19,25Temperature factors,
par-ticularly high in this protein region, experimentally corroborate such flexibility. The loop, numbered loop 7 in the (␣/)8 barrel, could trap the substrate by bringing the catalytic aspartic acid DPPA300 to an
attacking position facing the other catalytic residues
DPPA197 and EPPA233. Interestingly, a recent
crys-tallographic article, on human salivary ␣-amylase, probes sensible flexibility and evidences a role in substrate binding and enzyme activity for a short part of this loop 7, GHSA304 –HHSA310
26
(PPA number-ing and sequence similar in this part of the protein). A similar motion could also be hypothesized for barley loop 7 for HPPA305 has its HAMY2295 equivalent.
The crystallographic data concerning barley AMY2, either in its native or complex state, does not display an obvious “closed” or “open” position. However, a previous work27showed a very good superimposition
between oxygens of the crystallographic water mole-cules and oxygens (glycosidic or hydroxylic) of the modeled substrate. The water oxygens could mimic the substrate oxygens by sharing the same hydrogen-bond network, and thus could stick the loop in a closed position.27
The present article describes the characterization of this loop: identification, location, conservation, and especially motion. A careful examination of several ␣-amylases has listed and characterized all the struc-tural elements. The strucstruc-tural overview, combined with experimental evidence, identified loop 7 as flex-ible, part of the active cleft, and particularly con-served among␣-amylases. Then a thorough explora-tion of its putative moexplora-tion was necessary to highlight its dynamic role in the catalytic mechanism. The intrinsic motion and the structural consequences have been explored through a specific strategy called “ro-botics” where the suspected motion has been deliber-ately filtered and decomposed into a series of small displacements bordered by to-be-defined “open” and “closed” conformations. It corresponds to a peculiar constraint dynamics protocol. Since previous studies identified key residues of the active cleft for barley AMY2,27,28this methodology has been first applied to this enzyme. This work answers several questions, such as the following: What is the loop motion, its amplitude and its role? What are the consequences on catalysis: residues implied, trapping of transition state, or intermediate structures? Finally, what about a concerted motion?
METHOD
Loop 7: Identification, Conservation, and Flexibility
Identification Mode. TAKA, AMY2, and PPA
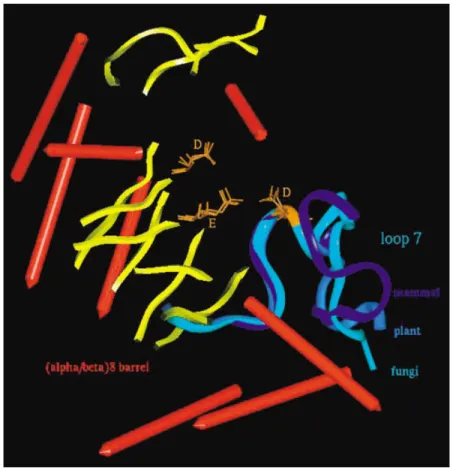
␣-amylases have been chosen to respectively repre-sent fungal, cereal, and mammal kingdoms. The su-perimposition mode is based on the backbone overlay of the strictly conserved catalytic triad composed of two aspartic acids and one glutamic acid. The archi-tecture of the catalytic site is known to be perfectly superimposed29 (Figure 1). To list all the flexible motifs that might be interesting in a dynamical ap-proach of the catalytic mechanism, the following set of criteria has been defined. All have to be rigorously satisfied: (1) a functional criterion, (2) a conservation criterion, and (3) a structural criterion.
The catalytic efficiency is expected to concern residues located in the active site, so the first criterion requires a location of the motif in the vicinity of the
catalytic triad. The second criterion requires for the motif the presence of some consensus sequence re-gion or highly conserved residues among␣-amylases. The third criterion, by strictly imposing a nonstruc-tural segment, discards automatically ␣-helix or -sheet described as rigid parts of a protein. Only loops satisfy this condition. To sum up, the identifi-cation of a putative catalytic flexible segment requires it to be a loop fragment, part of the active cleft, with both a primary sequence and a topography conserved within the family 13 of enzymes. According to these criteria, the loop 7 is the only one selected (Figure 1).
Protocol: Definition of two Barycenters and Flexi-bility Exploration of the Loop Without and With Substrates in the Active Site. The calculations are
performed on barley␣-amylase. The starting data are the crystallographic coordinates of the barley AMY2 complex (1bg9.pdb) with the pseudo-acarbose at 0.28 nm resolution.23,24 Since neither a substrate nor an inhibitor likely to limit or dodge the intrinsic motion of the loop is tolerated in the first stage, the acarbose inhibitor is removed from the active site. So in the first stage, the loop motion is explored without any substrate. In the second stage, to validate the ampli-tude of motion of the loop and explore its conse-quences on the hydrolytic mechanism, the motion of the loop is explored in the presence of a real substrate docked in the active site. Since molecular modeling is unable to simulate the cleavage of a covalent bond (only the quantum mechanics can theoretically answer this question and by now only quantum mechanics/ molecular mechanics coupling has approached a so-lution to this question30), the two main steps of the catalytic event, besides the proton transfer, are delib-erately chosen: the reaction steps before and after the cleavage. For the stage before the hydrolysis, a pre-viously modeled DP5 oligosaccharide31was chosen. Namely, the maltopentaose substrate Aⴚ1–A–B–C–D
is composed of glucose rings Aⴚ1, A, B, C, and D linked with a ␣-(1,4) bond from the nonreducing to the reducing end. It spans the active site from subsite ⫺2 to subsite ⫹3. For the stage after the hydrolysis, the loop underwent the same “robotics” strategy of motion in presence of maltose and maltotriose, prod-ucts resulting from the maltopentaose cleavage. The maltose Aⴚ1–A and maltotriose B–C–D occupy
re-spectively the subsites [⫺2; ⫺1] and [⫹1; ⫹3]. In all cases, for sake of CPU time, the enzyme is cut off before any calculations. The protein is re-stricted to the residues belonging to the loop, to its environment, to the subsites [⫺2; 3], plus the ones necessary for the structural integrity of the enzyme. All residues useless for calculations are discarded.
The molecular modeling is carried out on Silicon Graphics computers with the Accelrys packages (San Diego, CA, USA). Molecular displays and energy minimization are performed with Insight II, Biopoly-mer, and Discover modules. For all calculations, the CFF91 force field is selected.
Internal motions can be simulated by molecular dynamics calculations. Since loop motions are ex-pected to occur in the millisecond time scale, the required dynamics trajectory would have to be sam-pled in a period of time several times longer than a nanosecond. Moreover, such a long dynamics simu-lation would guarantee neither a complete description of the loop motion nor the evaluation of its amplitude. Actually, the suspected motion is one of medium frequency and could thus be delicate to extract from the high frequency multiple motions of a dynamic trajectory. Mouawad and Perahia have developed a method based on normal mode analysis.32,33By cou-pling vibration normal modes and Cartesian coordi-nates, low frequency motions were identified and characterized. Similarly, a method combining normal mode analysis and molecular dynamics simulation has been set up to analyze domain motions.34These
meth-ods are very useful but they are limited to small proteins or to large domain motions.
Due to the environment of this loop 7, the essential movement is a monodirectional back-and-forth motion with a peculiar shifting of the catalytic residue
DAMY2289 taking it closer to or further from its catalytic
counterparts DAMY2179 and EAMY2204 (Figure 2).
Be-cause the movement is decomposed into tiny steps sup-posed to be a relevant description of the reaction path, the procedure has been qualified as “robotics.” (Since it is an original description of the phenomenon, set up for this study, the quotations will be kept throughout the article.) In terms of calculations, the system is parti-tioned between rigid and mobile segments, where most of the shifting is supported by the loop left free to move. The constraints are thus supported by the remaining enzyme backbone beside the loop as well as the ends of the loop, which are residues VAMY2282 and SAMY2301
kept backbone fixed. This has been made possible with the definition of two barycenters (Figure 2):
1. B represents the rigid part and corresponds to the center of the mass between the catalytic
DAMY2179 and EAMY2204 backbone atoms.
FIGURE 1 Superimposition of TAKA (fungal), AMY2 (cereal), and PPA (mammal)␣-amylases, based on the backbone overlay of the strictly conserved catalytic triad.
2. Bⴕ represents the mobile loop and is defined from residues VAMY2282 to WAMY2297.
To give a rather continuous description of the phenomenon, the so-called “robotics” calculation is a succession of small displacements of Bⴕ toward B (and backward) under a distance constraint. Every distant restraint is followed by a significant 1500 iteration minimization to relax the internal coordi-nates of the protein. The goal of the procedure is to decrease the risk of false minima likely to occur with such a constraint trajectory essentially by relaxing the side-chain motion. Starting from the crystallographic position of the loop where the distance between B–Bⴕ is 1.52 nm, two trajectories are needed to describe the conformational space of the loop during its motion.
The two trajectories move from the x-ray position, set up arbitrarily to the medium position. The first one describes the loop flipping over the active site and the second one describes the loop flipping back to a totally accessible crevice. The two trajectories are then merged into a single one where the loop moves back and forth. The resulting trajectory is a succession of displacements of Bⴕ toward or away from B by steps of 0.02 nm on a range of more than 0.8 nm:
1. The first trajectory describes a shortening in the [B–Bⴕ] distance and brings DAMY2289 closer to
both EAMY2204 and DAMY2179 step by step.
This motion narrows the catalytic cleft and fi-nally leads to a completely closed position for loop 7. This artificial position is particularly FIGURE 2 Definition of the barycenters B and B⬘ and description of the robotics strategy.
much closer than the experimental x-ray con-formation that refers to the complex conforma-tion known to trap substrates or inhibitors. 2. The second trajectory corresponds to the
re-verse motion that enlarges the active cleft from the medium x-ray position to the “open” one. It corresponds to an increase in the B–Bⴕ distance, with DAMY2289 going further to the barycenters
(EAMY2204; DAMY2179). The broadening has
been fully explored so that the loop ends up in a completely open position.
The distance of 1.52 nm is artificially set to the relative value zero and the displacements towards the “closed” position are qualified with negative distance values; conversely the displacements towards the “open” position are noted as positive distance values (Figure 2). The experimental position, refined with a minimization similar to any step of the robotics strat-egy except the B–Bⴕ distance constraint, is obviously an acceptable geometry, so that its energy value can be taken as an energy threshold. Such a criterion borders the loop motion as it encompasses realistic conformations and discards odd ones at each extrem-ity. Any conformation beyond this energetic threshold is geometrically and energetically unbearable for the protein.
As mentioned above, the validation of the loop mo-tion as well as the comprehension of the different steps of the catalytic event imply the knowledge of the sub-strate behavior during the loop motion. For those rea-sons, a similar exploration is performed in the presence of a real substrate in the active site. Starting from the previously modeled maltopentaose27in the active site of barley␣-amylase with the loop this time in an “open” position, the “robotics” explores the motion of the loop until it reaches the crystallographic position supposed to be the very last step before the protonation of the cata-tytic bond. For a chronological continuity, the substrate is then artificially cleaved into maltose and maltotriose products known to occur from the hydrolysis of a mal-topentaose and the loop is then moved forward until it reaches a “closed” position.
RESULTS AND DISCUSSION Characterization of Loop 7
Identification of Loop on the Set of Functional and Biochemical Criteria. This superimposition displays
a close overlay of the structural elements of the (␣/)8
barrel where the eight helices and eight sheets have respectively their helical and strand equivalents. Only
loop 7, located between helix A-␣7 and sheet A-7 of the (␣/)8barrel, satisfies the three criteria previously
defined. Experimental data strongly support the flex-ibility of this loop 7 that flanks the active side and contains the fifth consensus region in ␣-amylases:
F–V–D–N–H–D. The last residue of this sequence is
the aspartic residue of the catalytic triad where the following numberings DTAKA298, DAMY2289, and DPPA300 refer respectively to TAKA, AMY2, and
PPA␣-amylases. It is worth mentioning that the three loops 7 show a backbone superimposition that goes largely beyond the consensus sequence and that gives a particularly low average RMS of 0.017 nm (Figure 1).
Main Structural Features of Loop 7. Despite a
ho-mologous folding for the part of the loop that corre-sponds also to the consensus sequence, the length of loop 7 varies with the␣-amylase origin. The TAKA amylase loop 7 has 10 amino acid residues (FTAKA292–FTAKA301), AMY2 has 15 residues
(FAMY2282–PAMY2300), and PPA 20 residues
(FPPA295–FPPA315). More generally, fungal and
bac-terial ␣-amylases display a particularly short loop with an average of 15 residues while cereal and mam-mal␣-amylases show a longer loop ranging from 15 to 25 residues.
Interestingly, some structural features provide sig-nificant internal rigidity to the mobile system. The loop joining sheet 7 and helix ␣7 has a typical ⍀ loop folding.9,10,35
1. The ⍀ loops are ⍀-shaped nonstructural seg-ments, located on the external side of globular proteins with a planar backbone.36 They are characterized by a lack of repetitive dihedral angles and of regular hydrogen bonds as well. However, intraloop hydrogen bonds and other elements such as turn are required to assure some rigidity to the loop and thus to guarantee its structural integrity during the motion. In cereal and mammal amylases, loop 7 adopts a typical ⍀ shape with a double turn at its top. Residues NAMY2287–GAMY2291 or NPPA298 – QPPA302 form the first turn that Kadziola
qual-ified, in barley, as␣7a.23The absence of such a turn, in the fungal amylase, is counterbalanced by a short twin loop parallel to loop 7. 2. Residue SAMY2292, located in the double turn,
shows (, ) values located in the forbidden region of the Ramachandran plot. It thus limits drastically the motion of both this residue and its neighbors.
3. Finally, the internal hydrogen-bond network for the three loops is dense and also participates in
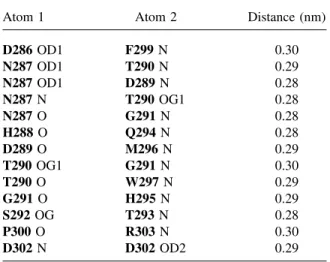
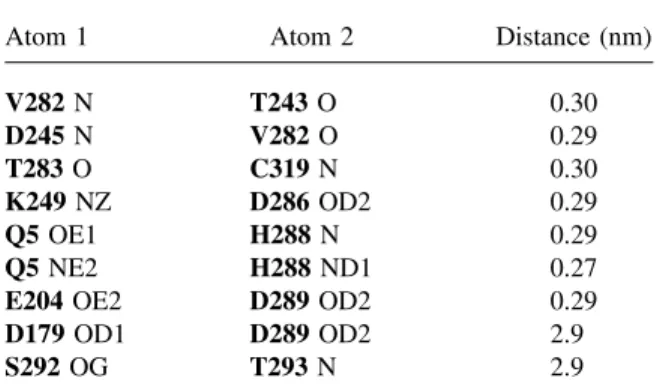
maintaining the general architecture of the loop. Tables I–VI list the intra- and interhydrogen bonds of AMY2 loop for the medium and ex-treme positions. The first three tables concern the intraloop hydrogen bonds from FAMY2284
to DAMY2302, the last three tables register the
interhydrogen bonds between loop 7 and its environment. In those tables, the residues in-volved will be seen as essential in the loop mobility despite a low hydrogen-bond number. The alternative solution of a poor hydrogen-bond network could be used in the case of a loop for which the folding flexibility would be particularly required. In the case of loop 7, the forbidden (, ) values of S292, the tight hydrogen-bond network combined to a ⍀-shape folding greatly favor the suspected rigid-body-type motion. This constrained folding is also consistent with a hinge-type motion where the hinge residues could be located at the bottom of the loop.
Environment of Loop 7. An accurate exploration of
the environment around loop 7 shows the presence of highly conserved aromatic residues: YAMY251 and WAMY29, YPPA62 and WPPA58, and YTAKA82.
TAKA-amylase lacks an equivalent WAMY29 or WPPA58 residue. A detailed spotting of AMY2
iden-tifies residues creating a hydrophobic upper rail on which leans the polar residue QAMY2294. WAMY29 is
actually sandwiched between QAMY2294 of loop 7
and QAMY249 so that its side chain is fixed in a given
conformation. This rail feature combined with the ⍀-shaped loop is fully coherent with the back and forth motion because QAMY2294 could slide along the
hydrophobic patch WAMY29. QAMY2294 could act as
a monitor to drive the loop forward and to bring
DAMY2289 closer to its catalytic DAMY2179 and EAMY2204 counterparts (Figure 1).
A final structural element is worth underlining as it could highlight some obscure points in the catalytic mechanism. Crystallographic data of TAKA, PPA, and AMY2 enzymes give water oxygens. Highly con-served oxygens have been shown to be located in a water pocket or channel adjacent to the active site. One wall of this water trap is formed with the con-served sequence F–V–D–N–H–D of loop 7. Kadziola suggested, in the case of AMY2, that the extreme water molecule of the channel (WAMY2607),
hydro-gen bridged between EAMY2204 and DAMY2289,
could act as the nucleophile during hydrolysis. The Table III Internal Hydrogen Bonds of Loop 7 in AMY2 in the “Open” Position
Atom 1 Atom 2 Distance (nm)
N287 N T290 OG1 0.30 Q294 NE2 D289 OD1 0.30 H295 NE2 S292 O 0.30 R303 N P300 O 0.29 V304 N S301 O 0.24 R303 NH1 D302 OD1 0.29 R303 NE D302 OD2 0.28
Table IV Hydrogen-Bond Network Between Loop 7 in the Crystallographic Position and Surroundings in AMY2
Atom 1 Atom 2 Distance (nm)
S292 OG W9 N 0.30
T293 OG1 G10 OE2 0.28
W297 O K249 NZ 0.29
W297 O N253 ND2 0.33
Table I Internal Hydrogen Bonds of Loop 7 in AMY2 in the Crystallographic Position
Atom 1 Atom 2 Distance (nm)
D286 OD1 F299 N 0.30 N287 OD1 T290 N 0.29 N287 OD1 D289 N 0.28 N287 N T290 OG1 0.28 N287 O G291 N 0.28 H288 O Q294 N 0.28 D289 O M296 N 0.29 T290 OG1 G291 N 0.30 T290 O W297 N 0.29 G291 O H295 N 0.29 S292 OG T293 N 0.28 P300 O R303 N 0.30 D302 N D302 OD2 0.29
Table II Internal Hydrogen Bonds of Loop 7 in AMY2 in the “Closed” Position
Atom 1 Atom 2 Distance (nm)
D286 OD2 T290 OG1 0.32 N287 OD1 T290 HN 0.30 N287 O G291 N 0.28 H288 O Q294 NE2 0.30 D289 OD1 Q294 NE2 0.30 W297 N T290 O 0.29 G291 O Q294 N 0.29 D302 OD1 R303 NH1 0.29 D302 OD2 R303 NE 0.28
loop motion, by implying a sensible motion of
DAMY2289, could trigger off the ejection of WAMY2607 towards the glycosidic linkage and thus
could drive the water molecule to the attacking posi-tion expected in the second step of the reacposi-tion mech-anism. In that way, the loop motion could not only enhance the turnover of the enzyme but could also favor the water supply of this channel or pocket.
Flexibility of Loop 7.
Energetic Diagram and Amplitude Motion When the Active Site Is Empty. Molecular mechanics protocol adopted a deliberately filtered “robotics” analysis. The successive variations of Bⴕ displacements are considered as discrete snapshots of the loop motion and a sequential description of the reaction pathway. All the positions after minimization are merged into a unique trajectory file where each [B–Bⴕ] distance corresponds to a total potential energy value. The total potential energy variation versus [B–Bⴕ] distance is plotted in Figure 3.
The diagram shows a rather symmetrical curve with energy increases for the two extreme positions and interestingly the medium crystallographic one. The energy plot confirms a back-and-forth motion as acceptable. According to the energetic criterion, three positions are discriminated: the crystallographic one (the medium position) and the two extreme positions (“closed” and “open”) bordering out the loop dis-placement, respectively located at ⫺0.24 nm and ⫹ 0.4 nm from the crystallographic position; the former value corresponds to a “closed” position, the latter to an “open” one. Despite a rather rough energy criterion, this bordering allows a large amplitude of motion of more than 0.6 nm.
Key Points of the Loop Motion. Generally speaking, the trajectory tracks down the conformational reaction path of the loop during its motion. The extreme and intermediate positions seen in Figure 3 show that the
⍀ shape is kept all along the loop motion, thus con-firming the architectural importance of the ⍀ loop characteristics previously underlined.
Crystallographic Position. The crystallographic
conformation and position of the loop is red in Figure 3. Interestingly, the energy plot displays a peak at this position. As it is a refined experimental conformation, a minimum in potential energy should be expected. Among the different hypotheses that can explain the original shape of the curve, one could argue some “memory effect” of the inhibitor due to the initial coordinates of the complex AMY2/acarbose. Even if the robotics calculations demand that the inhibitor should be discarded, residues that used to be in con-tact with the inhibitor could have maintained their side chains in specific geometry difficult to relax. The second hypothesis relates also to the acarbose inhib-itor that is supposed to mimic the transition state. This suggests that the enzyme is deluded and adopts an activated conformation, thus increasing its conforma-tional energy. In all cases, the loop in this x-ray and intermediate position would initiate the distortion of the substrate towards its transition state.
To elucidate this tricky point, a continuous and reverse loop motion is performed completely from the newly defined “closed” position toward the “open” one. A similar strategy is used: same distance range, same calculation protocol (step of 0.02 nm for each [B–Bⴕ] distance variation, 1500 iteration minimiza-tion under similar constraints). This second trajectory no longer displays an energy increase at the corre-sponding crystallographic position. The energy plot shows a regular curve with maxima at the extreme positions and minimum at the medium crystallo-graphic position. This robotics trajectory confirms once more the possibility of a back-and-forth motion for loop 7. The increase observed in the previous trajectory at the crystallographic position could be due to a kind of memory effect for the protein that used to have its crevice occupied with the acarbose inhibitor.
Extreme Geometries.
The opened position. The opened position is
shown in green in Figure 3. The energy diagram Table V Hydrogen-Bond Network Between Loop 7 in
the “Closed” Position and Surroundings in AMY2
Atom 1 Atom 2 Distance (nm)
V282 N T243 O 0.30 D245 N V282 O 0.29 T283 O C319 N 0.30 K249 NZ D286 OD2 0.29 Q5 OE1 H288 N 0.29 Q5 NE2 H288 ND1 0.27
E204 OE2 D289 OD2 0.29
D179 OD1 D289 OD2 2.9
S292 OG T293 N 2.9
Table VI Hydrogen-Bond Network Between Loop 7 in the “Open” Position and Surroundings in AMY2
Atom 1 Atom 2 Distance (nm)
V282 N FT243 O 0.30
D245 N V282 O 0.28
C319 N T283 O 0.29
K249 OZ2 V285 O 0.29
shows a putative motion of 0.4 nm from the starting crystallographic position without noticeable steric conflict. Despite no measurement performed in terms of surface accessibility, the catalytic cleft is obviously enlarged. The broadening of the cleft while the loop flips back to an open position could facilitate the arrival of substrate, particularly the helical one. This capability to dock a helical chain of substrate should confirm the efficiency of barley amylases on native solid substrate. These enzymes are actually known to act on starch granules during the physiological con-ditions of germination where amylose and amylopec-tin chains are folded in helical conformations. This result could be a step toward the understanding of amylolysis in heterogeneous phase condition.
As the loop flips over the active site, some of its residues could be implied in the early stage of sub-strate docking. Several residues involved in subsite binding belong to loop 7, especially HAMY2288, DAMY2289, and MAMY2296 that have been
identi-fied27,28as critical for the docking of short or for long substrates. Among residues that also define the cata-lytic crevice, YAMY2104 and segment [EAMY2204 – KAMY2216] from loop 5 could act in synergy with the
loop 7 and thus partially disrupt the helical structure of the amylose chain. Through strengthened interac-tions, the first steps of the substrate docking could be facilitated.
The closed position. The so-called “closed”
con-formation corresponds to the yellow position in Fig-FIGURE 3 Energy diagram (energy plot vs [B–Bⴕ] distance) and amplitude motion of the loop
ure 3. The diagram displays putative motion ampli-tude of 0.25 nm between the medium and the close positions. The energy variations during the loop mo-tion is too delicate to interpret qualitatively; however, the large increase in energy, beyond the cutoff, can obviously be explained by steric clashes between res-idues of the loop within its environment.
We propose the following chronological and cata-lytic process as the loop moves from the intermediate position to the “closed” one:
First, the dynamical rearrangement of the residues of the crevice results in a tighter binding of the sub-strate and more generally in a correct positioning of the reacting agents.
Then the negatively charged DAMY2289 moves
closer to the substrate. On the other side of the cleft, the protonated EAMY2204, now at a catalytic distance
from the reacting sugars, is able to transfer its proton to the glycosidic oxygen, thus promoting the distor-tion of the glucose ring and the formadistor-tion of the transition state.
Finally, the loop motion could promote the ejection of a water molecule from the water channel and could place it in an attacking position. This point is partic-ularly interesting as the water molecule could act as the nucleophile in the reaction process.
In a further chronological step while the loop keeps on moving in, some cooperative side-chain reposi-tioning between the loop residues and their environ-ment could favor the reaction product release. This point will be particularly illustrated in the paragraph of the loop motion in the presence of products.
Loop Characteristics in the Presence of Substrate. To validate the loop flexibility, we studied the ampli-tude of the loop motion and its functional conse-quences when the active site is filled with substrates. This “robotics” study actually requires two trajecto-ries. The first (part I) describes the maltopentaose behavior when the loop moves from an “open” state to the crystallographic position; then the second trajec-tory (part II) describes the behavior of the products when the loop moves from the RX position to the “closed” position (Figure 4, upper). The expected result is a functional comprehension of the loop mo-tion. The first trajectory shows how the loop motion acts realistically both for the tight docking and for the distortion of the substrate towards its transition state geometry while the second trajectory displays a dy-namic release of the products once the hydrolysis is done.
Loop Motion with the Maltopentaose. During the
trajectory, specific frames have been deliberately se-lected as they describe very specific steps of the
reactional path. The images in Figure 4 (lower right) correspond respectively to
● a completely open loop (green loop),
● the half open position and the minimum in en-ergy (light green), and
● the RX position (red loop).
The maltopentaose Aⴚ1–A–B–C–D occupies
sub-sitesⴚ2 to ⴙ3 so that the glucose ring A is docked in subsite ⴚ1 where the catalysis occurs. This ring is highly suspected to go through a distorted half-boat conformation during the catalysis. The set of Pucker polar parameters (Q,⌰,) characterize the distortion of a glucose conformation where the4C1chair
con-formation is the reference for a ␣-D glucose ring.
Table VII shows the evolution of the Pucker param-eters for the glucose ring A during the loop motion. Actually, as the loop moves forward, the substrate gets trapped tighter by stacking interactions between ring A and Y51 and between ring C and W206. Then, as the loop goes on moving in, the ring A in subsite ⴚ1 undergoes a significant distortion measured with the ⌰ parameters going from 11.95° to 23.7°. The distortion, results in a final half-boat H2conformation
for ring A known to be the transition state conforma-tion. The transition from the4C1conformation to the
H2 conformation is helped by the residues H288, D204, and D289 (Figure 4, lower right).
Loop Motion with the Maltotriose and Maltose.
During the trajectory, some specific frames are selected as they interestingly illustrate the release of the products during the reactional path. When the loop flips over the active site, numerous specific interactions between the sugar and the protein are disrupted, thus weakening the docking of the sub-strate. In fact, during the closure of the crevice, residues YAMY251, HAMY2288, WAMY2206, MAMY2296, and WAMY2297 accommodate their
side chain so as to reduce the steric hindrance. During their repositioning, they unstick respec-tively from rings A, B, C, and D (Figure 4, lower left: on this figure for sake of clarity, only the unstick of the maltotriose B–C–D is shown). In-deed, some loop residues compete with glucose rings to create stacking interactions with aromatic residues. Actually, two pairs of amino acid resi-dues: HAMY2288/YAMY251 and MAMY2296/ WAMY2206 illustrate this putative property (Figure
4, lower left and right). The first pair of residues is a highly conserved set. What is more, YAMY251 and WAMY2206 are referenced as conserved stacking
patches37–39for substrate glucose rings.27,28During the loop closure, HAMY2288 residue moves in and
creates a hydrogen bond with the side chain of
YAMY251, thus inducing a relevant reorientation of
the tyrosine side chain. Such competing interaction weakens the affinity between YAMY251 and the
glucose ring A and could be responsible for the disruption of the stacking interaction. Similarly, the significant displacement of MAMY2296 residue
ini-tiates a significant displacement of WAMY2206 side
chain, thus decreasing the binding affinity between glucose ring C and the stacking subsite WAMY2206.
Hinge Residues. An overview of protein motions
showed that the intrinsic flexibility of some segments can be classified into hinge or shear motions.1In our case, the back and forth motion should be obviously compared to the first-type-hinge-motion as seen for the TIM ⍀ loop.6 – 8 Despite neither significant nor very located variation in the C␣ dihedral values, the ⍀ shape of the (␣/)8 barrel loop 7 combined with a
pivotal displacement of the HAMY2288 and DAMY2289 residues are strong structural features
FIGURE 4 Upper: chronological and functional comprehension of the loop motion. Lower right: distortion of the maltopentaose at subsiteⴚ1 to get to its transition state geometry. Lower left: release of the cleaved substrate, details of the leaving maltotriose on the reducing end.
which suggest a hinge-type motion. Indeed, one could expect that residues located at the bottom could act as lever arm residues and a small variation in their (, ) values will induce a significant displacement of resi-dues located at the top. Interestingly, those resiresi-dues are the catalytic DAMY2289 and the important HAMY2288 and MAMY2296 residues. A thorough
ex-ploration identified VAMY2285 and FAMY2299 as
pu-tative hinge residues with VAMY2285 interestingly
belonging to the fifth consensus sequence FVDNHD. The two residues are located at the bottom sides of the ⍀ loop where the loop gets narrower. They are both hydrophobic residues connected with van der Waals interaction and with their side chain always in tight contact during the loop motion.
More generally, a comparison between AMY2,
TAKA, and PPA ␣-amylases revealed a similar
“gear” mechanism with a conserved set of hinge res-idues located in the same area of the ⍀ loop. In TAKA-amylase, the hinge pair is strictly conserved with VTAKA293 and FTAKA301 residues, in PPA
amylase, VPPA296 is strictly conserved while FAMY2299 is replaced by a similarly hydrophobic
isoleucine IPPA313.
CONCLUSIONS
This work is a first step toward a structural and functional three-dimensional mapping of␣-amylases. The comparison of␣-amylases from different origins combined crystallographic data, consensus sequence superimposition, and structural and functional fea-tures identification. Criteria have been set up here to select regions that are interesting as flexible and func-tional motifs. The criteria require a nonfolded seg-ment (structural criterion) that contains a consensus sequence (biochemical criterion) and that flanks the active site (functional criterion).
From a survey of ␣-amylases and data selected from fungal (TAKA), cereal (AMY2), and mammal (PPA, HSA) amylases, a loop has been identified as potentially flexible and strongly involved in the amylolysis. Loop 7 has been proved here to be the
loop among the eight ones of the TIM barrel that satisfies the structural, functional, and biochemical criteria. Among the␣-amylases, all the loops 7 dis-play a characteristic⍀-shape loop, referenced as flex-ible in many TIM barrel proteins with different func-tion. This⍀ loop is enriched with a strong internal hydrogen-bond network, with a double turn on top, and in the case of AMY2, with forbidden (, ) values in the Ramachandran plot for a serine residue. It induces some internal rigidity for the loop but this tenseness has been shown to be consistent with the suspected back and forth movement. A detailed ex-ploration of AMY2 loop 7 surroundings shows a rail that could support and monitor the loop in its motion. This rail is formed of other loops of the TIM barrel, generally speaking, most of the loops could deserve a study but the paper here focused on loop 7.
Barley AMY2 whose crevice was already charac-terized in terms of subsite residues27, 28was chosen to
explore the putative catalytic loop 7. An original robotics strategy has been developed where the very high and very low frequency motions were deliber-ately filtered to get into the loop motion of medium frequency. As a back-and-forth motion was suspected, loop 7 has been successively moved in and out of its crystallographic position considered as an intermedi-ate one. The loop flexibility has been validintermedi-ated with a similar “robotics” strategy in the presence of substrate or products of hydrolysis. In all cases, the calculations explored the mobility of the loop and bordered its motion with a precise identification of “close” and “open” conformations. The loop displacement showed a non-negligible amplitude of more than 0.6 nm; a 0.4 nm displacement from the “open” to the medium positions and a 0.24 nm displacement from the medium to the “closed” positions.
From this study, the motion could be seen as a cooperative displacement of residues, initiated by hinge residues located at the bottom of the loop. A small displacement of the pivotal VAMY2285 and FAMY2299 could trigger the loop movement.
Consis-tently, a similar set of “hinge” residues has been found highly conserved in␣-amylases. The two res-Table VII Pucker Parameters Evolution for Ring A Docked in Subsite -1
Loop Position, Energy Open, Maximum Intermediate, Intermediate Half Open, Minimum Intermediate, Intermediate RX, Maximum Q 0.542 0.541 0.542 0.542 0.542 ⌰ 11.95 14.23 16.05 20.00 23.68 74.85 75.68 77.30 83.56 89.59
idues are now proposed as rational mutation points to validate or not the hinge hypothesis.
A chronological scheme of the loop motion is proposed with extreme conformations corresponding to key steps in the adsorption/desorption mechanism. The “open” conformation obviously enlarges the cleft so that the adsorption of substrate, even the native helical one, is largely facilitated. When the loop flips over, the residues of the loop such as HAMY2288, QAMY2294, and MAMY2296 move forward in synergy
with their crevice counterparts, respectively
YAMY251, WAMY29, and WAMY2206 to reinforce the
substrate binding, to promote the transition state for-mation and then to position DAMY2289 to an attacking
position for catalysis. Located in the close vicinity of the glycosidic linkage and the protonated catalytic
EAMY2204 residue, DAMY2289 along with DAMY2179
could stabilize the transition state through a covalent intermediate after the departure of the leaving group. The ultimate closure of the loop considerably de-creases the width of the cleft and enhances coopera-tive side-chain motions of residues YAMY251/ HAMY2288 and WAMY2206/MAMY2296, thus
favor-ing the release of the cleaved fragments. In a final step, desorption of the products is thus largely fa-vored.
In the calculations, the water molecules have been deliberately discarded. It is known that ␣-amylases display a water pocket in the vicinity of the active site where one side of the pocket is formed by loop 7. Those water molecules are highly suspected to act as nucleophiles. The crystallographic data showed the exiting water molecule of the pocket involved in bridging EAMY2204 and DAMY2289. One could
sus-pect that the displacement of the loop would position the water molecule as the attacking nucleophile. A further understanding of the AMY2 loop would help to analyze the dynamic behavior of the water mole-cules during the loop motion. These calculations would give a more realistic energetic pathway for the loop motion with some insight on the actual supply of water molecules as nucleophiles. As loop 7 could be considered as a structural and a functional motif, an ultimate work would be a research of such conserved motifs combined with a dynamic exploration on other amylases of the family 13.
REFERENCES
1. Gerstein, M.; Lesk, A. M.; Chotia, C. Biochemistry 1994, 33, 22, 6739 – 6749.
2. Arteca, A. A. Biopolymers 1996, 39, 671– 680.
3. Van Vlijmen, H. W. T.; Karplus, M. J Mol Biol 1997, 267, 975–1001.
4. Brzovic, P. S.; Hyde, C. C.; Miles, E. W.; Dunn, M. F. Biochemistry 1993, 10404 –10413.
5. Andre´, G.; Kanchanawong, P.; Palma, R.; Cho, H.; Deng, X.; Irwin, D.; Himmel, M. E.; Wilson, D. B.; Brady J. W. Protein Eng 2003, 16, 125–134.
6. Joseph, D.; Petsko, G. A.; Karplus, M. Science 1990, 249, 1425–1428.
7. Rhee, S.; Parris, K. D.; Hyde, C. C.; Ahmed, S. A.; Miles, E. W.; Davies, D. R, Biochemistry 1997, 36, 7664 –7680.
8. McMillan, M. F.; Cahoon, M.; White, A.; Hedstrom, L.; Petsko, G.; Ringe, D, Biochemistry 2000, 39, 4533– 4542.
9. Ring, C. S.; Kneller, D. G.; Langridge, R.; Cohen, F. E. J. Mol. Biol 1992, 224, 685– 699.
10. Fetrow, J. S. FASEB J 1995, 9, 708 –717.
11. Matsuura, Y.; Kusunoki, M.; Harada, W.; Kakudo, M. J Biochem 1984, 95, 697–702.
12. Brzozowski, A. M.; Davies, G. J. Biochemistry 1997, 36, 10837–10845.
13. Boel, E.; Brady, L.; Brzozowski, A. M.; Derewenda, Z.; Dodson, G. G.; Jensen, V. J.; Petersen, S. B.; Swift, H.; Thim, L.; Woldike, H. F. Biochemistry 1990, 29, 6244 – 6249.
14. Machius, M.; Wiegand, G.; Huber, R. J Mol Biol 1995, 246, 545–559.
15. Fujimoto, Z.; Takase, K.; Doui, N.; Momma, M.; Ma-tsumoto, T.; Mizuno, H. J Mol Biol 1998, 277, 393– 407.
16. Aghajari, N.; Feller, G.; Gerday, C.; Haser, R. Protein Sci 1998, 7, 564 –572.
17. Strobl, S.; Maskos, K.; Betz, M.; Wiegand, G.; Huber, R.; Gomis-Ru¨th, F. X.; Glockshuber, R. J Mol Biol 1998, 278, 617– 628.
18. Qian, M.; Haser, R.; Payan, F. J Mol Biol 1993, 231, 785–799.
19. Qian, M.; Haser, R.; Buisson, G.; Due´e, E.; Payan, F. Biochemistry 1994, 33, 6284 – 6289.
20. Brayer, G. D.; Luo, Y.;Withers, S. G. Protein Sci 1995, 4, 1730 –1742.
21. Ramasubbu, N.; Bhandary, K. K.; Scannapieco, F. A.; Levine, M. J. Proteins Struct Funct Genet 1991, 11, 230 –232.
22. Ramasubbu, N.; Paloth, V.; Luo, Y.; Brayer, G. D.; Levine, M. J. Acta Crystallogr 1996, D52, 435– 446. 23. Kadziola, A.; Abe, J.; Svensson, B.; Haser, R. J Mol
Biol 1994, 239, 104 –121.
24. Kadziola, A.; Søgaard, M.; Svensson, B.; Haser, R. J Mol Biol 1998, 278, 205–217.
25. Gilles, C.; Astier, J. P.; Marchis-Mouren, G.; Cambil-lau, C.; Payan, F. Eur J Biochem 1996, 238, 561–569. 26. Ramasubbu, N.; Ragunath, C.; Mishra, P. J. J Mol Biol
2003, 325, 1061–1076.
27. Andre´, G.; Bule´on, A.; Haser, R.; Tran, V. Biopolymers 1999, 50, 751–762.
28. Andre´, G.; Tran, V. Special Publication No. 246, The Royal Society of Chemistry; Gilbert, H. J., Davies, G. J., Henrissat, B., Svensson, B., Eds.; Cambridge, 1999 pp 165–174.
29. Uitdehaag, J. C. M.; Mosi, R.; Kalk, K. H.; van der Veen, B. A.; Dijkhuizen, L.; Withers, S. G.; Dijkstra, B. Nature Struct Biol 1999, 6, 432– 436.
30. Gao, J. In Reviews in Computational Chemistry: Methods and Applications of Combined Quantum Mechanical and Molecular Mechanical Potentials; Lipkowitz, K. B., Boyd, D. B., Eds.; VCH Publishers: New York, 1996. 31. Andre´, G.; Bule´on, A.; Juy, M.; Aghajari, N.; Haser, R.;
Tran, V. Biopolymers 1999, 49, 107–119.
32. Mouawad, L.; Perahia, D. Biopolymers 1993, 33, 299 – 611.
33. Mouawad, L.; Perahia, D. J Mol Biol 1996, 258, 393– 410.
34. Hayward, S.; Kitao, A.; Berendsen, H. J. C. Proteins Struct Funct Genet 1997, 27, 425– 437.
35. Leszczynski, J. F.; Rose, G. D. Science 1986, 234, 849 – 855.
36. Fetrow, J. S.; Spitzer, J. S.; Gilden, B. M.; Scott, J. M.; Begley, T. J.; Haas, B. J.; Boose, T. Biochemistry 1998, 37, 2477–2487.
37. Quiocho, F. A. Ann Rev. Biochem 1986, 55, 287– 315.
38. Quiocho, F. A. Pure Appl Chem 1989, 61, 1293–1306. 39. Vyas, N. K. Curr Opin Struct Biol 1991,1, 732–740. 40. Bak-Jensen, K.; Andre´, G.; Tran, V.; Svensson, B.
J Biol Chem, to be published.