Étude du dynamisme et de l'évolution des réseaux
d'interactions protéiques par une approche de
protéomique comparative
Mémoire
Caroline Berger
Maîtrise en biologie - avec mémoire
Maître ès sciences (M. Sc.)
Étude du dynamisme et de l’évolution des
réseaux d’interactions protéiques par une
approche de protéomique comparative
Mémoire
Caroline Berger
Sous la direction de :
Christian Landry
Résumé
Un objectif fondamental de la biologie évolutive est de comprendre comment l’information contenue dans le génotype peut être transmise au phénotype. Les hybrides, issus du croisement entre deux espèces différentes, représentent une opportunité unique d’explorer le lien qui existe entre génotype et phénotype. L’hybridation peut mener à la mise en place de phénotypes extrêmes (tels que l’hétérosis ou la sous-dominance) et beaucoup d’études s’intéressent aux bases génétiques de ces phénotypes. Pourtant, il y a une véritable lacune dans notre compréhension du lien entre génotype et phénotype chez les hybrides. Notre hypothèse était que ce lien se fait par l’intermédiaire des complexes protéiques et que l’hybridation devrait induire une réorganisation des complexes. Pour tester cette hypothèse, nous avons utilisé une méthode d’étude des complexes protéiques (SEC-PCP-SILAC) qui permet de cibler un grand nombre de complexes dans la cellule. Des hybrides de levures ont été générés au laboratoire et SEC-PCP-SILAC a été utilisé pour comparer les complexes protéiques des hybrides par rapport aux complexes parentaux. Nous avons été en mesure de capturer une large fraction de l’interactome avec la détection de 39% des complexes protéiques présents chez la levure. Nos résultats mettent en évidence la robustesse générale des complexes protéiques après hybridation. Toutefois, des modifications non négligeables du réseau d’interactions ont aussi été détectées. Ces modifications affectent deux voies biologiques majeures : la voie de synthèse du glucose et la voie liée à l’activité ribosomale. Ce sont des voies candidates intéressantes pour expliquer la différence de phénotype qui peut exister entre parents et hybrides. Finalement, l’utilisation d’une méthode alternative, la PCA, a permis de complémenter les données de spectrométrie de masse, en démontrant notamment la présence d’interactions parentales (intra-espèces) et chimériques (inter-espèces) chez les hybrides. Ce mémoire souligne ainsi l’importance d’adopter une approche intégrative pour une meilleure compréhension du lien génotype-phénotype.
Abstract
A fundamental goal in evolutionary biology is to understand how the information contains in the genotype can be transmitted to the phenotype. Hybrids, that are the result of the cross between different species, represent a unique opportunity to investigate the link between genotype and phenotype. Hybridization can lead to extreme phenotypes (such as heterosis or underdominance) and many studies try to understand the genetic bases of these phenotypes. However, there is a real gap in our understanding of the link between genotype and phenotype in hybrids. Our hypothesis was that protein complexes would play a key role and that hybridization would lead to changes in the organisation of protein complexes. To test this hypothesis, we used a method (SEC-PCP-SILAC) that allows studying broadly protein complexes in the cell. Hybrids between yeast species were generated in the laboratory and SEC-PCP-SILAC was applied to compare the protein complexes of the hybrids with their parental species. We were able to detect a large fraction of the interactome with the identification of 39% of the protein complexes reported in yeast. Our results highlight the general robustness of the protein complexes after hybridization. However, some significant changes of the interaction networks were also detected in hybrids. These modifications involve two main biological pathways: the glucose synthesis pathway and the ribosomal activity pathway. They are promising candidates to explain the phenotypic differences between hybrids and parents. Finally, a complementary PCA approach was used to complement the mass-spectrometry data and we demonstrated the presence of both parental (within species) and chimeric (between species) interactions. This thesis emphasizes the importance to use an integrative approach for a better understanding of the link between genotype and phenotype.
Table des matières
Résumé ... ii
Abstract ... iii
Table des matières ... iv
Liste des figures ... vi
Liste des tableaux ... vii
Liste des abréviations, sigles, acronymes ... viii
Remerciements ... xiv
Avant-propos ... xvi
Introduction ... 1
Du génotype au phénotype ... 1
Premières descriptions de la carte génotype-phénotype ... 3
La biologie des systèmes ... 4
Étude des réseaux d’interactions protéiques : perturbation ... 7
L’hybridation entre espèces comme modèle pour l’étude des interactions protéiques ... 10
Les phénotypes extrêmes des hybrides ... 10
Les bases génétiques de la sous-dominance ... 11
Les bases génétiques de l’hétérosis ... 13
Le lien manquant entre génotype et phénotype : réseaux d’interactions protéiques .. 14
Méthodes d’étude des réseaux d’interactions protéiques ... 17
Détection des interactions physiques entre protéines par complémentation fonctionnelle ... 17
Détection des complexes protéiques par purification par affinité et spectrométrie de masse ... 19
Incertitudes de ces méthodes : faux-positifs et faux-négatifs ... 21
Émergence de nouvelles méthodes : SEC-PCP-SILAC ... 23
Objectif du projet ... 24
Chapitre 1: Comparative proteomics highlights the general robustness of protein complexes after hybridization ... 27
1.1 Résumé ... 28
1.2 Abstract ... 29
1.3 Introduction ... 30
1.4 Results ... 34
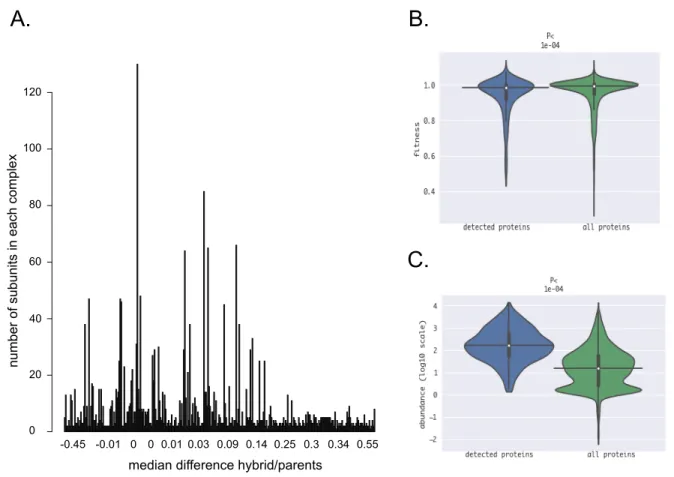
1.4.1 The SEC-PCP-SILAC method allows detection of a large fraction of the interactome in yeast strains ... 34
1.4.2 Protein complexes show general robustness in hybrids ... 38
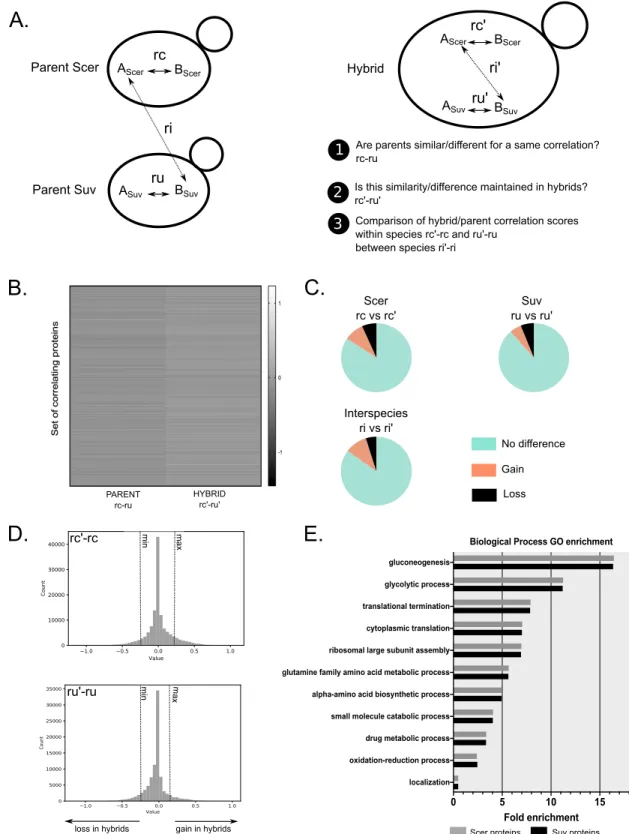
1.4.3 Comparison of correlation scores highlights differences between parents and hybrids ... 41
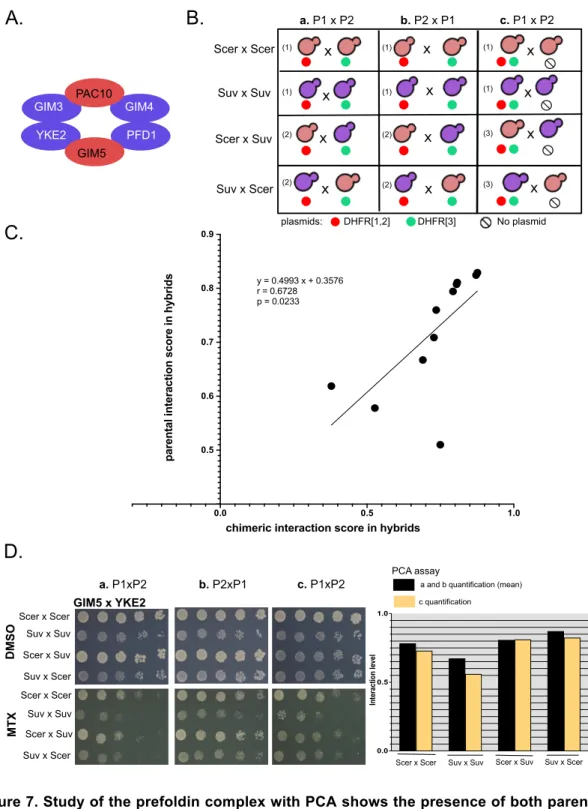
1.4.4 Complementation of the mass spectrometry results with another method ... 44
1.5 Discussion ... 47
1.6 Conclusion ... 50
1.7 Materials and Methods ... 50
1.7.1 Generation of yeast hybrids ... 50
1.7.2 SILAC labelling ... 51
1.7.3 Cell lysis ... 51
1.7.4 Size-Exclusion Chromatography (SEC) ... 52
1.7.5 Protein digestion ... 52
1.7.6 Mass spectrometry ... 53
1.7.7 Database searching and quantification ... 54
1.7.8 Reference databases of protein complexes ... 54
1.7.9 Estimation of correlation scores ... 55
1.7.10 Comparison between parents and hybrids ... 55
1.7.11 Construction of strains for the DHFR-PCA in the prefoldin complex ... 56
1.7.12 Screening of PPIs in the prefoldin complex ... 57
1.7.13 PCA images and quantification ... 58
1.8 Supporting information ... 59
1.8.1 Supplementary figures ... 59
1.8.2 Supplementary tables ... 63
Conclusion ... 67
Liste des figures
Figure I. Les réseaux d’interactions protéiques occupent une place centrale au sein des
cartes génotype-phénotype ... 6
Figure II. Les interactions entre espèces perturbent le réseau d’interactions protéiques ... 8 Figure III. Phylogénie du genre Saccharomyces chez la levure ... 25 Figure 1. Scenario of the evolution of protein complexes after inter-species hybridization ... 31 Figure 2. Workflow of the SEC-PCP-SILAC method used for the detection of the protein
complexes ... 35
Figure 3. SEC-PCP-SILAC allows detection of a large fraction of the interactome in
parental and hybrid yeast species ... 37
Figure 4. The vast majority of protein complexes detected with the SEC-PCP-SILAC
method shows robustness after hybridization ... 39
Figure 5. Several features might explain the general robustness of protein complexes .. 41 Figure 6. Analysis of correlation scores allows detection of differences between hybrids
and parents in specific pathways ... 43
Figure 7. Study of the prefoldin complex with PCA shows the presence of both parental
and chimeric interactions in hybrids ... 46
Figure S1. Experimental optimization of the SEC-PCP-SILAC method in yeast parental
and hybrid species ... 59
Figure S2. Bioinformatic workflow applied for the identification of protein complexes in
yeast parental strains and their hybrids using SEC-PCP-SILAC ... 60
Figure S3. The two parental strains Scer and Suv are divergent species ... 61 Figure S4. Additional interactions used for detection of chimeric and parental interactions
Liste des tableaux
Table S1. Description of the crosses performed in this study:
Table S1A. Crosses performed in the SEC-PCP-SILAC experiment ... 63
Table S1B. Crosses performed in the DHFR-PCA experiment ... 63
Table S2. SILAC labelling combinations used during the SEC-PCP-SILAC experiment . 64 Table S3. Results of the SEC-PCP-SILAC experiment (unique and all analysis): Table S3A. List of identified protein complexes ... 64
Table S3B. Elution profiles of individual proteins: results of parents all analysis ... 64
Table S3C. Elution profiles of individual proteins: results of hybrids all analysis ... 64
Table S3D. Elution profiles of individual proteins: results of parents unique analysis ... 64
Table S3E. Elution profiles of individual proteins: results of hybrids unique analysis ... 64
Table S4. List of the proteins assigned to strong changes in hybrids compared to parents: Table S4A. Proteins assigned to Scer sequences ... 64
Table S4B. Proteins assigned to Suv sequences ... 64
Table S5. Primers used in this study ... 65
Table S6. List of the protein complexes used as references for the identification of the protein complexes in the SEC-PCP-SILAC experiment ... 66
Liste des abréviations, sigles, acronymes
% : Pourcentage°C : Degré Celsius
A600 : Absorbance d’un échantillon mesurée pour une longueur d’onde de 600 nm ADN : Acide désoxyribonucléique
AP-MS : Affinity Purification and Mass Spectrometry (Purification d’Affinité et Spectrométrie de Masse)
ARN : Acide ribonucléique
BCA : BiCinchoninic acid Assay (Test à l’Acide BiCinchoninique)
BN-PAGE : Blue Native Polyacrylamide Gel Electrophoresis (Électrophorèse sur Gel Bleu Natif de Polyacrylamide)
BRET : Bioluminescent Resonance Energy Transfer (Transfert d’Énergie de Résonance de Bioluminescence)
ClonNAT : Nourséothricine cm : Centrimètres
Da : Dalton
DBH: Dosage Balance Hypothesis (Hypothèse de la Balance de Dose) DHFR : Dihydrofolate réductase
DMSO : Diméthylsulfoxyde dNTP : Désoxyribonucléotides DTT : Dithiothréitol
ESI : Electrospray Ionization (Ionisation par Électronébuliseur) eV: électronvolt
FDR : False Discovery Rate (Taux d’erreur)
FRET : Fluorescence Resonance Energy Transfer (Transfert d’Énergie entre Molécules Fluorescentes)
GO : Terme de Gene Ontology
H Lysine (or H sample) : Heavy Lysine (Lys8) HCl : Acide chlorhydrique
Hz : Hertz
IAA : Iodoacetamide
IEX : Ion-Exchange Chromatography (Chromatographie à Échange d’Ions) IgG : Immunoglobulines
IQR: Interquartile Range (Écart Interquartile) KCl: Chlorure de potassium
L : Litre
L lysine (or L sample) : Light Lysine (Lys0) LD : Limite de Détection
LOWESS : Locally Weighted Scatterplot Smoothing (Méthode de régression locale) Lys : Lysine
m : Mètre
M : Concentration molaire (mole par litre) M lysine (or M sample): Medium Lysine (Lys4) MATa : Type sexuel a
MAT𝛼 : Type sexuel alpha min : Minute
mL : Millilitre mg : Milligramme
MgCl2 : Chlorure de magnésium
mM : Millimolaire
MS : Mass Spectrometry (Spectrométrie de Masse) MS/MS : Spectrométrie de masse en tandem
MTX : Méthotrexate
MWCO : Molecular weight cut off (seuil de poids moléculaire) m/z: Rapport masse/charge
NaCl : Chlorure de sodium NaoAc : Acétate de sodium NaoH : Hydroxyde de sodium nm : Nanomètres
pb : Paire de bases
PCA : Protein-fragment Complementation Assay (Test de Complémentation de fragments de Proéines)
PCNA : Proliferating Cell Nuclear Antigen (Antigène Nucléaire de Prolifération Cellulaire)
PCP : Protein Correlation Profiling (Profil de Corrélation des Protéines) PCR : Polymerase Chain Reaction (Réaction en Chaîne par Polymérase)
PIN : Protein Interaction Network (Réseau d’Interactions Protéiques) PPI : Protein-Protein Interaction (Interaction Protéine-Protéine)
PTM : Post Translational Modification (Modification Post-Traductionelle) ppm : Partie par millions
Q1 : Quartile 1 Q3 : Quartile 3
QTL : Quantitative Trait Loci (Locus de Caractères Quantitatifs) r : Coefficient de corrélation
RNApol : ARN polymérase rpm : Rotations Par Minute s : Seconde
Scer : Saccharomyces cerevisiae
SDS-PAGE : Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (Électrophorèse en Gel de Polyacrylamide contenant du Dodécylsulfate de Sodium)
SEC : Size-Exclusion Chromatography (Chromatographie d’Exclusion Stérique) SILAC : Stable Isotope Labeling by Amino acids in Cell culture (Marquage Istotopique Stable par les Acides Aminés en Culture Cellulaire)
Suv : Saccharomyces uvarum
TAP : Tandem Affinity Purification (Purification par Affinité en Tandem) TBS: Tris-Buffered Saline
TEV: Tobacco Etch Virus (Virus de la Gravure du Tabac) TFA: Acide Trifluoroacétique
Th: Thomson (unité) V: Volt
Y2H : Yeast Two-Hybrid (Double Hybride) YPD : Yeast extract, Peptone, Dextrose µg : Microgramme
µL : Microlitre µm : Micromètre µM : Micromolaire
[...] et la morale de ceci, c'est : Soyez ce que vous voudriez avoir l'air d'être ;
ou, pour parler plus simplement : Ne vous imaginez pas être différente de ce qu'il eût pu sembler à autrui que vous fussiez ou eussiez pu être en restant identique à ce que vous fûtes sans jamais paraître autre que vous n'étiez avant d'être devenue ce que vous êtes.
Lewis Carroll,
Remerciements
Ce mémoire est l’aboutissement de quatre années passées au sein du laboratoire de Christian Landry et j’aimerais donc remercier toutes les personnes qui ont contribué à cet accomplissement. Tout d’abord, j’aimerais remercier mon directeur de recherche, Christian. Merci de m’avoir confié ce beau projet. C’était quelque chose de tout nouveau pour le laboratoire alors il y a forcément eu des choses qui ont marché, d’autres qui ont moins bien marché, mais j’ai eu une vision complète de la recherche avec ses hauts et ses bas! Merci de m’avoir fait confiance pour ce projet. Tu as été un formateur hors pair tout au long de mon parcours, tu as toujours été là pour me conseiller et m’orienter même dans les moments où j’avais l’impression de me perdre. J’ai énormément appris au cours de ces quatre années, non seulement sur le plan scientifique, mais aussi sur moi-même. Je pars de ton laboratoire avec une plus grande confiance en moi et la certitude du chemin que je veux suivre. Merci pour tout cela.
J’aimerais remercier Steve Charette, qui a été co-directeur pendant une partie de mon projet. La co-direction s’est arrêtée avec la fin du projet sur les paramécies, et pourtant Steve a continué de me suivre tout au long de mon parcours. Merci Steve d’avoir toujours su me prêter une oreille attentive. Merci de tes conseils qui ont été très précieux pour moi. Merci à Isabelle Gagnon-Arsenault et Alexandre Dubé, deux personnes qui ont été particulièrement importantes pour moi au cours de ces quatre années. Vous êtes tous les deux passionnés par votre travail et vous savez transmettre cette passion aux personnes qui vous entourent. Merci pour votre aide, pour votre bienveillance et vos conseils, qui, j’en suis certaine, me seront utiles bien au-delà des murs d’un laboratoire.
Plusieurs personnes ont contribué à ce projet, et j’aimerais les remercier : merci à Rohan Dandage, qui est stagiaire postdoctoral au sein du laboratoire de Christian et qui a effectué les analyses bio-informatiques des données de spectrométrie de masse. Tu as fait un travail exceptionnel et notre bonne entente se reflète dans le papier final. Ce projet était aussi une collaboration avec le laboratoire de Léonard Foster, de l’Université de Colombie-Britannique. J’ai eu la chance de faire un stage de cinq mois dans le laboratoire de Léonard. Merci à l’ensemble du laboratoire de Léonard pour leur accueil chaleureux. Merci Léonard de m’avoir guidé dans les toutes premières étapes de ce projet. Merci à Jenny Moon pour m’avoir aidé dans les étapes expérimentales et pour avoir pris soin de
mes nombreux échantillons après mon départ! Finalement, merci à Greg Stacey, bioninformaticien du laboratoire de Léonard, qui nous a conseillé pour la mise en place de l’expérience, et plus tard pour l’analyse des données.
Je tiens également à remercier les membres de mon comité d’encadrement : Steve Charette, Nicolas Bisson et Nicolas Derome. Vos conseils m’ont beaucoup apporté et ont été très utiles au projet. Merci à Steve Charette et Nicolas Bisson qui ont accepté de prendre du temps pour évaluer mon mémoire de maîtrise. Votre expertise et avis seront très appréciés.
Merci à Julie Turgeon, directrice du programme de maîtrise en biologie, qui a été à mon écoute et qui a su me guider et m’aider au cours de certaines étapes de mon parcours. Merci à l’ensemble des membres du laboratoire Landry, qui était une petite famille à mon arrivée, qui est devenue une plus grande famille au moment de mon départ! Merci pour nos discussions et nos échanges, qu’ils soient ou non d’ordre scientifique! J’aimerais remercier certaines personnes en particulier : merci à Souhir Marsit, Axelle Marchant, Clara Bleuven et Éléonore Durand. Merci d’avoir été des amies présentes pour moi, merci pour nos discussions, votre support, et les bons moments passés ensemble. Merci également à Carla et Angel pour votre gentillesse et votre bonne humeur quotidienne. Finalement, j’aimerais remercier les personnes sans qui tout cela n’aurait pas été possible : ma famille. Merci à ma maman, Monique, qui a été là chaque seconde de mon parcours et qui n’a jamais cessé de croire en moi. Merci pour tous les cadeaux et sacrifices que tu as a faits avec papa pour assurer à tes filles un bel avenir. J’ai notamment appris de toi la force et le courage et tu seras toujours un modèle à suivre pour moi. Merci à ma sœur jumelle Chloé qui est à mes côtés à chaque instant. Les expériences que nous avons vécues au cours des dernières années n’ont fait que renforcer un lien indéfectible. Tu sais combien tu comptes pour moi. Merci maman et Chloé d’avoir été ma lumière au cours de ces dernières années. Je ne cesserai jamais de la suivre.
Je pense de tout mon cœur aux personnes que j’aime, mais qui ne sont malheureusement plus là. Merci à mon papa Gérard, à mon papi Roger, et à ma mamie Renée. Vous êtes à chaque instant dans mon cœur.
Avant-propos
Ce mémoire comporte un unique chapitre, rédigé sous la forme d’un article scientifique qui sera soumis pour publication au cours de l’année 2020. Cet article porte sur l’étude de l’évolution des complexes protéiques après hybridation entre espèces, en utilisant une méthode émergente de protéomique comparative (SEC-PCP-SILAC). Je suis co-première auteure de cet article, avec Rohan Dandage. J’ai contribué à la planification des expériences avec Christian R. Landry, Léonard J. Foster et Isabelle Gagnon-Arsenault. Isabelle Gagnon-Arsenault, Kyung-Mee Moon et moi-même avons participé à l’exécution des expériences. Isabelle Gagnon-Arsenault et moi-même avons construit les souches mentionnées dans le projet (expériences SEC-PCP-SILAC et PCA). J’ai préparé l’ensemble des échantillons pour l’expérience SEC-PCP-SILAC. Kyung-Mee Moon a réalisé les manipulations impliquant le spectromètre de masse incluant les recherches avec le logiciel MaxQuant. Isabelle Gagnon-Arsenault et moi-même avons effectué l’expérience et l’analyse des données PCA. L’analyse des données brutes de spectrométrie de masse (analyse des profils d’élution et établissement des complexes protéiques) a été réalisée par Rohan Dandage, qui est indiqué comme co-premier auteur dans l’article. Les analyses bio-informatiques subséquentes de comparaison entre hybrides et parents ont été réalisées par Rohan Dandage et moi-même. Richard Greg Stacey nous a conseillé dans la planification de l’expérience et dans les stratégies d’analyse bio-informatique. J’ai rédigé le manuscrit de l’article présenté en chapitre 1, avec la contribution de Christian R. Landry et Rohan Dandage.
Introduction
Du génotype au phénotype
Un des grands défis de la biologie actuelle est de comprendre le lien qui existe entre génotype et phénotype (Balaresque & King, 2016; Feigin & Mallarino, 2018; Martin, 2015; Peichel & Marques, 2017). Au sens large, le terme génotype désigne l’ensemble des gènes d’un organisme. Dans un sens plus restreint, le terme peut être utilisé pour les allèles, ou formes variantes d’un gène, portés par un organisme (Johansen, 1909; Roll-Hansen, 2009). Le terme phénotype désigne quant à lui les propriétés physiques observables d’un organisme. Le phénotype d’un organisme est influencé par son génotype, mais aussi par les facteurs environnementaux (Borders, 1971; Churchill, 1974). Des exemples de phénotypes comprennent la longueur des ailes, la couleur des poils, ou encore des caractéristiques mesurables en laboratoire telles que les taux d’hormones. Les récentes découvertes et innovations en science et technologie ont permis l’accès à des milliers de génomes séquencés et un grand nombre de gènes ont été caractérisés (Heather & Chain, 2016; Maschke, 2016). En parallèle, nous sommes de nos jours en mesure de décrire un grand nombre de phénotypes, de la variation naturelle à la maladie. Par exemple, la base de données « Online Mendelian Inheritance in Man » (OMIM) référence de nombreuses maladies humaines (Amberger & Hamosh, 2017). Malgré cet accroissement dans l’acquisition des données, il y a une grande lacune dans notre compréhension du lien entre génotype et phénotype. Pour de nombreuses maladies, comprendre leurs bases génétiques n’a pas conduit à un traitement efficace à cause de cette insuffisance dans nos connaissances. Par exemple, nous savons que le syndrome de Down est causé par la présence d'une copie supplémentaire du chromosome 21, mais nous ne comprenons pas pourquoi les individus atteints ont des symptômes variables allant du retard mental à des anomalies faciales, et nous ne savons pas pourquoi le phénotype varie de léger à extrêmement sévère (Papavassiliou, Charalsawadi, Rafferty, & Jackson-Cook, 2015).
Plusieurs raisons peuvent expliquer cette lacune dans notre compréhension du lien entre génotype et phénotype. Tout d’abord, connaître le phénotype total (c'est-à-dire l'ensemble des phénotypes ou « phénome ») d'un individu demanderait de l'observer avec tous les outils d'analyse possibles et dans toutes les conditions environnementales possibles
(Hebbring, 2014). Ensuite, les quelques vingt-deux milles gènes humains ne suffisent certainement pas aux innombrables caractères phénotypiques d'un humain : le génotype ne produit qu’en partie le phénotype. Une fraction importante du phénotype sera
également déterminée par des mécanismes épigénétiques, c’est-à-dire des mécanismes
modifiant de manière réversible, transmissible et adaptative l'expression des gènes sans changement de séquence nucléotidique. Ces changements épigénétiques incluent la méthylation de l’ADN, les modifications d’histone, les ARN non codants et la structure de la chromatine (Gayon, 2016; Kelly & Issa, 2017). La situation est d’autant plus complexe dans la mesure où l’environnement peut avoir une influence sur le phénotype (Thessen et al., 2015). Par exemple, le rayonnement solaire peut influencer les mécanismes impliqués dans la synthèse d'un pigment sombre, la mélanine (Pillaiyar, Manickam, & Jung, 2017). Le mémoire présenté ici a pour objectif global de mieux comprendre ce lien complexe qui existe entre génotype et phénotype. Comment l’information contenue dans le génotype est-elle transmise au phénotype ? Par quels mécanismes des modifications de l’information contenue dans le génotype peuvent mener à des variations du phénotype ? Pour répondre à ces questions, nous reviendrons dans un premier temps sur un bref historique de la compréhension de la carte génotype-phénotype. Cet historique nous mènera à la représentation actuelle de la carte génotype-phénotype, qui place les réseaux d’interactions protéiques au centre de la carte. Nous verrons que l’hybridation entre espèces, en perturbant ces réseaux, est un modèle très prometteur pour l’étude de la relation génotype-phénotype. L’introduction mettra finalement la lumière sur les méthodes utilisées pour étudier les réseaux d’interactions protéiques, et la nécessité de développer de nouveaux outils permettant l’étude des réseaux d’interactions protéiques à grande échelle. Le chapitre 1 sera présenté sous forme d’article scientifique. Ce chapitre illustrera comment l’étude de l’hybridation entre espèces et l’utilisation d’une méthode récente de spectrométrie de masse peuvent conduire à une meilleure compréhension du lien génotype-phénotype.
Premières descriptions de la carte génotype-phénotype
La question des mécanismes menant aux variations phénotypiques n’est pas récente. Dès 1868, Darwin observe des structures hypothétiques localisées au sein des cellules. Il appelle ces structures des « gemmules » et émet l’hypothèse qu’elles sont responsables des variations phénotypiques et de leur hérédité (Darwin, 1868). Quelques années plus tard, Mendel, père de la génétique, démontre le concept de gène récessif et de gène dominant : il propose que des « facteurs invisibles » sont responsables des caractères et de leur hérédité. Le terme de gène n’est certes pas encore mis en place à l’époque, mais Mendel fut l’un des premiers à faire la distinction entre génotype et phénotype (Mendel, 1936). Ce n’est que des années plus tard que la première relation directe entre génotype-phénotype fut établie.
En 1902, Garrod établit pour la première fois la relation entre un gène et une enzyme grâce à l'étude de patients souffrant d'alcaptonurie. Cette maladie se transmet comme un facteur mendélien récessif et est attribuable à l'absence d'une enzyme de la dernière étape du métabolisme de la tyrosine. Il fait alors le rapprochement entre gène et enzyme, mais cette hypothèse ne peut cependant pas être formellement démontrée à cause de la complexité des voies métaboliques correspondantes (Garrod, 1902). En 1941, Beadle associé à Tatum, apportent une démonstration de la relation « un gène = une enzyme » en étudiant un organisme plus simple : le champignon Neurospora crassa. Ils utilisent une méthode qui consiste à réaliser l'analyse biochimique et génétique des mutants déficients pour une voie de synthèse de l’arginine. En associant systématiquement un gène mutant à l'incapacité d'effectuer une réaction métabolique donnée, ils parviennent à montrer qu'à un gène correspond une enzyme (Beadle & Tatum, 1941). On est alors en mesure de démontrer qu’un caractère est exprimé à partir d’un gène et qu’une variation de la séquence de ce gène peut induire une variation du caractère qui lui est associé. C’est l’ébauche d’une première carte génotype-phénotype.
Le courant réductionniste apparaît alors. On pense qu’un gène est associé à un caractère
individuel et que les organismes et leurs phénotypes résultent en la somme de ces associations. Comprendre comment fonctionne un organisme reviendrait à comprendre la fonction spécifique et le caractère associés à chacun des gènes qui le constituent (Jacob, 1970). L’organisme est perçu comme une véritable « machine organique », par analogie avec une machine industrielle. Le canard mécanique de Vaucanson est un des exemples
pour lesquels le comportement animal est « réduit » et comparé à l’effet d’un mécanisme physique (Moran, 2007).
Il apparaît aujourd’hui évident que la vision réductionniste présente plusieurs limites. Le modèle réductionniste s’appuie sur le lien simple qui existe entre un gène et un caractère. Par exemple, une mutation dans le gène contrôlant la synthèse de l’hémoglobine HBB induit l'apparition de globules rouges rigides de forme allongée chez les personnes souffrant d’anémie falciforme (Strouse, 2016). La vision réductionniste établit ainsi une relation causale entre génotype et phénotype, soit l’élimination d’un gène qui résulte en un phénotype, ce qui permet de déterminer la fonction de ce gène. Cependant on doit être prudent avec cette relation de causalité. Un cœur produit un son de battement, mais cela ne veut pas dire que la fonction principale du cœur est de produire du son. Un segment d’ADN peut être transcrit en ARN, mais beaucoup reste à faire pour démontrer que cette transcription a un rôle du point de vue fonctionnel. De plus, de nombreux caractères complexes résultent de l’expression non pas d’un unique gène, mais de plusieurs gènes. Waddington fut un des premiers à proposer en 1952 que le phénotype des organismes résulte en fait d’interactions complexes entre les gènes (Waddington, 1952).
Ainsi, la vision réductionniste appliquée au monde du vivant rencontrait de nombreuses difficultés, attribuables principalement à la complexité des interactions dans les systèmes vivants. Ces limites ont conduit à de nouvelles approches et méthodes pour une meilleure compréhension du lien entre génotype et phénotype.
La biologie des systèmes
La notion de système est apparue en biologie dans les années 50, proposée par Von Bertalanffy, un biologiste dont l’approche servira de fondement à la théorie générale des systèmes (Von Bertalanffy, 1951). Un système est défini comme un ensemble d’éléments interagissant entre eux selon des règles particulières. Il est déterminé par la nature de ses composants, leurs interactions, et sa frontière, c’est-à-dire les éléments essentiels à son fonctionnement. Un système peut être ouvert s’il interagit avec son environnement, ou fermé, et peut être composé de sous-systèmes.
Dans ce contexte, la biologie des systèmes étudie les relations et les interactions entre les différentes parties du système biologique : organites, cellules, systèmes physiologiques, réseaux de gènes ou réseaux de protéines (Kitano, 2002). Les organismes vivants ne sont plus comparés à des machines (comme c’était le cas dans l’approche réductionniste), mais sont considérés comme des systèmes ouverts, composés de sous-systèmes imbriqués les uns dans les autres. Les systèmes vivants, contrairement aux systèmes mécaniques artificiels, vont avoir des caractéristiques uniques telles que leur capacité d’auto-organisation puisqu’ils contiennent l’information et les outils nécessaires à celle-ci (Neumann, 1966).
La vision du lien entre génotype et phénotype devient alors plus complexe. On considère que les différents réseaux moléculaires (réseaux d’interactions protéine-protéine, réseaux de régulation des gènes, réseaux métaboliques, etc.) forment des couches intermédiaires entre le génotype et le phénotype (Diss et al., 2013; Tucker, Gera, & Uetz, 2001) (Figure I). Parmi ces différentes couches, ce sont les réseaux d’interactions protéiques (ou interactome) qui jouent un rôle central dans la connexion entre génotype et phénotype. Les protéines s’associent ensemble pour réaliser la plupart des fonctions dans la cellule et pour transmettre l’information entre l’environnement, le génome, et la cellule (Diss et al., 2013; J. D. Scott & Pawson, 2009). Ainsi, l’étude des interactions protéine-protéine apparaît comme une stratégie d’analyse prometteuse puisqu’on est alors en mesurer d’aborder la problématique du lien génotype-phénotype à un niveau hiérarchique moins complexe. Il n’en reste pas moins que l’interactome n’est qu’une pièce du puzzle et que seule l’intégration de toutes les pièces (épigénome, transcriptome, métabolome, RNAome, modifications post-traductionnelles) permettra à long terme une vision plus exhaustive du lien entre génotype et phénotype (Figure I).
PTM code
METABOLOME
GENOME
EPIGENOME
RNAome
INTERACTOME
DEVELOPMENT TIME EVOLUTION ENVIRONMENT INFECTIONFigure I. Les réseaux d’interactions protéiques occupent une place centrale au sein des cartes génotype-phénotype. L’intégration des informations du génome, de l’épigénome, du
transcriptome, du métabolome, du protéome, du RNAome et des modifications post-traductionnelles (PTM Post-Translational Modifications) permettra de comprendre comment la cellule répond à des facteurs externes tels que l’environnement, l’interaction avec d’autres organismes, le temps de développement et la durée de processus physiologiques tels que le cycle cellulaire ou l’évolution. Adapté de Diss et al. 2013.
Étude des réseaux d’interactions protéiques : perturbation
L’étude des réseaux d’interactions protéiques peut reposer sur le postulat que c’est en perturbant les interactions entre protéines (PPI Protein-Protein Interactions), et donc l’architecture des réseaux d’interactions protéiques (PIN Protein Interaction Networks), que les variations génotypiques mènent à des variations phénotypiques (Tucker et al., 2001; Vidal, Cusick, & Barabási, 2011). Ainsi, la compréhension du lien entre génotype et phénotype peut se faire en induisant une perturbation des réseaux d’interactions protéiques. Tel que reporté par Diss et al., les perturbations générées peuvent être de différents types (Diss et al., 2013).
Une des manières les plus intuitives d’induire une modification du réseau d’interactions protéiques est par mutation. Lorsqu’un gène est muté dans une séquence codante, les modifications d’acides aminés peuvent affecter les interactions d’une ou plusieurs protéines, aboutissant à une modification de l’architecture des PIN (Matija et al., 2009). Par exemple, Zhong et al. ont étudié les mutations associées à des maladies humaines et ont identifié les changements de PPI induits par ces mutations. Ils ont démontré que des mutations distinctes dans un même gène (par exemple TP63) peuvent générer différents phénotypes en fonction des PPI perturbées (Zhong et al., 2009). Dans une autre étude, Diss et al. ont étudié l’interaction de protéines dupliquées dans un contexte sauvage et dans un contexte de délétion de l’une des deux copies. Ils ont démontré que 22 des 56 paires à l’étude compensaient la perte d’interactions. Un nombre équivalent de paires démontrait le comportement contraire puisque la présence des deux copies était nécessaire pour maintenir leurs interactions. Ces résultats sont une autre illustration du fait que des mutations distinctes (ici des délétions) peuvent modifier les PIN et induire des phénotypes différents (fragilité ou robustesse) (Diss et al., 2017).
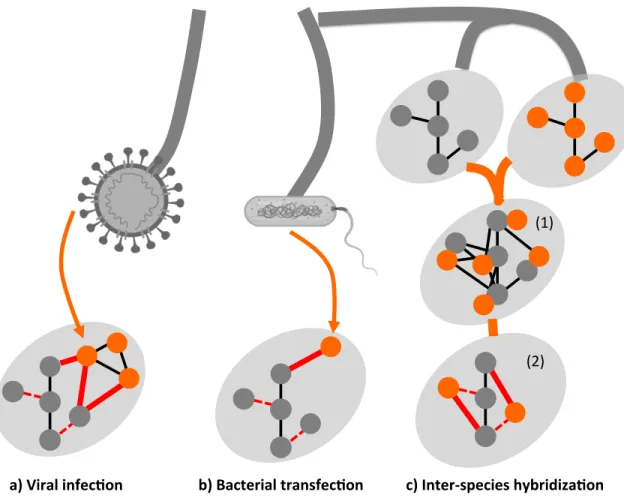
En plus des mutations, d’autres mécanismes peuvent induire une modification des PIN. Les interactions entre espèces représentent une source majeure de perturbation et sont donc une voie prometteuse pour l’étude des réseaux d’interactions protéiques (Diss et al., 2013). On parle ici d’évènements qui conduisent au contact moléculaire entre deux organismes et à l’interaction entre leurs PIN mutuels (Figure II).
a) Viral infec,on b) Bacterial transfec,on c) Inter-species hybridiza,on
(1)
(2)
Figure II. Les interactions entre espèces perturbent les réseaux d’interactions protéiques. a) Les infections virales (branche de gauche) entraînent un contact entre les composants des
réseaux protéiques de l’hôte et du virus. Les interactions entre les protéines virales et les protéines de l’hôte (lignes épaisses rouges) vont perturber l’interactome de l’hôte (lignes pointillées rouges) et donc les processus cellulaires. b) Dans le cas des infections bactériennes (centre), l’interactome de l’hôte sera perturbé par des protéines effectrices de la bactérie, plutôt que par l’intégralité de l’interactome de la bactérie (ligne épaisse rouge). Ces effecteurs peuvent également modifier l’interactome de l’hôte (lignes pointillées rouges). c) Lorsqu’il y a hybridation entre deux espèces distinctes (branche de droite), il y a un mélange initial des deux interactomes parentaux (étape 1). Après hybridation (étape 2), l’interactome de l’hybride sera composé d’interactions inter-espèces dites chimériques (lignes rouges) et d’interactions intra-espèces dites parentales (lignes noires). Certaines interactions auront été perturbées (lignes pointillées rouges), alors que d’autres apparaissent (lignes épaisses rouges). Adapté de (Diss et al., 2013).
Les interactions hôte-pathogène, comme celles qui ont lieu entre bactéries, virus et leurs cellules hôtes sont un bon exemple de ce type d’interactions (Mukhtar et al., 2011; Orit et al., 2012; Stefanie et al., 2011). La longue coévolution entre les agents pathogènes et leur hôte devrait favoriser les agents pathogènes qui réussissent à manipuler leur hôte en ciblant des protéines clés du réseau d’interaction de leur hôte. Ces PPI deviennent alors des candidats potentiels pour les traitements thérapeutiques contre les maladies infectieuses. Par exemple, Mukhtar et al. ont analysé comment des protéines (effecteurs) produites par des bactéries pathogènes affectaient les PPI de la plante Arabidopsis thaliana. Ils ont démontré que les effecteurs agissaient au niveau des protéines les plus
connectées (le hub ou centre du réseau) du PIN (Mukhtar et al., 2011). Des résultats similaires ont été obtenus pour les virus (Andreas et al., 2012; A. C. Michael et al., 2007). De nombreux défis demeurent toutefois. Premièrement, il reste à étudier le lien entre PPI de l’hôte et de l’agent pathogène dans leur contexte biologique. Ceci est particulièrement important dans les cas où les protéines effectrices pathogènes sont modifiées dans le contexte cellulaire de l’hôte (Selbach et al., 2009). Deuxièmement, une autre voie prometteuse consistera à comprendre le rôle des interactions inter-espèces dans des situations non pathogènes. Il serait par exemple intéressant de tester si des stratégies similaires de ciblage du noyau (hub) des PIN sont utilisées par des virus et des bactéries, dans des contextes d’interactions mutualistes ou endosymbiotiques.
Le modèle hôte-pathogène reste un modèle émergent et relativement peu d’informations sont disponibles en ce qui concerne le génotype et le phénotype des pathogènes et de leur hôte. Les agents pathogènes sont souvent difficiles à garder en laboratoire, difficiles à reproduire et à faire croître en quantité suffisante, et ils parasitent des hôtes qui ne sont pas des sujets expérimentaux idéaux. De nombreux agents pathogènes, tels que Plasmodium vivax, ne peuvent pas être maintenus en culture. Ce n’est que récemment
que les progrès récents dans le domaine du séquençage ont permis d’obtenir la séquence de plusieurs espèces parasites telles que Plasmodium falciparum (Bartholomeu & El-Sayed, 2004; Gilabert et al., 2018; Winzeler, 2009).
L’hybridation entre espèces comme modèle pour l’étude des
interactions protéiques
Un autre cas extrême de perturbation du réseau d’interactions protéiques dans le cadre d’interactions entre espèces est celui des hybrides. Un hybride provient du croisement de deux espèces différentes. Le modèle des hybrides, contrairement à celui hôte-pathogène, est très documenté et plusieurs informations sont disponibles sur leurs phénotypes et leurs génotypes (Anderson & Stebbins, 1954; Barton, 2001; Dittrich-Reed & Fitzpatrick, 2013).
Les phénotypes extrêmes des hybrides
Concernant le phénotype des hybrides, la première hypothèse qui peut s’imposer est que l’hybride montrera un phénotype intermédiaire à celui des parents. Cependant, de nombreuses études montrent aujourd'hui que le phénotype hybride peut être extrême (Bar-Zvi, Lupo, Levy, & Barkai, 2017). Plusieurs cas de sous-dominance ont été rapportés, situation pour laquelle le phénotype des hybrides est inférieur à celui des parents (Maheshwari & Barbash, 2011). Par exemple, les hybrides entre le riz indica et le riz japonica cultivés en Asie sont stériles (Ouyang, Liu, & Zhang, 2010). L’autre cas de phénotype extrême chez les hybrides survient lorsque le phénotype des hybrides est supérieur à celui des parents : c’est ce que l’on appelle l’hétérosis (Lippman & Zamir, 2007). Par exemple, les hybrides entre le faisan de Colchide (Phasianus colchicus) et les poules domestiques (Gallus gallus) sont plus gros que leurs parents (Darwin, 1868). Cette caractéristique est très exploitée dans l’industrie agroalimentaire puisque les plants hybrides montreront un meilleur rendement. Un exemple économiquement important est le maïs hybride qui offre un avantage considérable sur le rendement en semences par rapport aux espèces parentales. Les semences hybrides dominent le marché commercial des semences de maïs aux États-Unis, au Canada et dans de nombreux autres grands pays producteurs de maïs (Smith, Betrán, & Runge, 2004).
De nombreux efforts ont été déployés pour comprendre les bases génétiques à l’origine des différents phénotypes hybrides (dans un contexte de recherche fondamentale, mais aussi probablement dans un contexte économique).
Les bases génétiques de la sous-dominance
Selon Charles Darwin, la sous-dominance des hybrides n'est pas un produit de la sélection naturelle. Il écrit que le phénomène résulterait de la divergence des espèces qui s'hybrident, plutôt que de l'action directement exercée par des pressions sélectives (Darwin, 1859). Les études contemporaines ont confirmé la compréhension de Darwin. Quelles sont donc les bases génétiques de la sous-dominance?
Les changements de ploïdie (c’est-à-dire du nombre de chromosomes) semblent un facteur important de sous-dominance observée chez les hybrides de plantes, bien que ce soit moins le cas chez les hybrides d’animaux (Beukeboom, Koevoets, Morales, & Ferber, 2015; Coyne & Orr, 2004). Les réarrangements chromosomiques contribuent également à la stérilité hybride et à la non-viabilité chez les animaux et les plantes. Cependant, des évidences de plus en plus nombreuses indiquent que la sous-dominance hybride est en grande partie le résultat d'allèles situés à différents loci génétiques qui sont « incompatibles » (Johnson, 2000). Chez la drosophile, seuls quelques gènes, caractérisés et séquencés, contribueraient à la sous-dominance des hybrides (Chung & Chau-Ti, 2004).
La première avancée majeure dans la compréhension des bases génétiques de la sous-dominance des hybrides est le modèle de Bateson-Dobzhansky-Muller, qui résulte d’une combinaison de résultats de William Bateson en 1909, et de Theodosius Dobzhansky et Joseph Muller entre 1937 et 1942 (Bateson, 1909; Dobzhansky, 1951; Muller, 1942). Le modèle explique pourquoi un effet négatif sur le phénotype, tel qu’observé dans les cas de sous-dominance, ne sera pas éliminé de la population par sélection négative. Le modèle stipule que la sous-dominance est très probablement provoquée par la fixation alternative de deux loci ou plus, au lieu d'un seul locus, de sorte que, lorsque l'hybridation se produit, certains allèles s’expriment pour la première fois chez un même individu. Par exemple, imaginons deux populations récemment séparées géographiquement. Les deux populations ont initialement le même génotype AABB. Une population peut alors évoluer vers aaBB (via une étape intermédiaire AaBB), tandis que l'autre population évolue vers AAbb (via une étape intermédiaire AABb). Au cours de ces processus, l’allèle a sera en présence des allèles A et B, et l’allèle b sera également exprimé avec les allèles A et B, sans conséquence sur le fitness des organismes. Lorsque l'hybridation se produit, c'est la première fois que les allèles a et b s’expriment ensemble, l’interaction entre ces deux allèles est incompatible. Un exemple du modèle de Bateson-Dobzhansky-Muller est
observé chez des poissons (Coyne & Orr, 2004). De nombreux individus du poisson platy Xiphophorus maculatus ont des taches sur leurs nageoires dorsales, tandis qu'une espèce proche (le poisson porte-glaive X. helleri) montre l’absence de taches (Coyne & Orr, 2004). Chez certains hybrides rétrocroisés de ces espèces, les taches sont plus grandes et se développent en tumeurs malignes, ce qui réduit la durée de vie des organismes. En fait, les poissons platy avec des taches ont un gène lié au chromosome X qui produit des taches, et tous les poissons platy ont un répresseur autosomal qui vérifie l'expression du gène produisant des taches. En revanche, les poissons porte-glaive sont dépourvus à la fois du gène producteur de taches et du répresseur. Dans les rétrocroisements, certains des hybrides reçoivent le gène producteur de taches, mais pas le répresseur. Ces individus sont ceux qui développent des tumeurs malignes, car l'expression du gène producteur de taches n'est pas correctement régulée. Ce modèle peut s’appliquer à une bien plus large échelle. Des études ont démontré que des hybrides de Drosophila étaient stériles en raison de l’interaction d’une centaine de gènes (Wu, Johnson, & Palopoli, 1996).
Les avancées technologiques des dernières années ont permis d’aller plus loin dans le modèle de Bateson-Dobzhansky-Muller. Par exemple, le modèle « snowball effect » (effet boule de neige) prédit une accumulation de loci incompatibles au fil du temps. Comme le modèle de Bateson-Dobzhansky-Muller postule que la stérilité est attribuable à une interaction allélique négative entre les espèces qui s'hybrident, au fur et à mesure que les espèces divergent, le nombre d’incompatibilités augmentera de manière exponentielle (Gourbière & Mallet, 2010). Il faut aussi garder en tête que, bien qu'une multitude de preuves soutiennent le modèle de Bateson-Dobzhansky-Muller, cela n'exclut pas la possibilité que d'autres situations génétiques puissent conduire à une sous-dominance des hybrides. Par exemple, Lynch et Force ont proposé que lorsque la duplication de gènes se produit, il est possible qu'un gène redondant soit rendu non fonctionnel au fil du temps par des mutations. Une population pourra perdre la fonction de la première copie, alors que l’autre population perdra la fonction de la seconde copie. Les hybrides résultant pourraient n’avoir aucun gène fonctionnel issu de la paire dupliquée (Lynch & Force, 2000). L'héritage épigénétique pourrait aussi contribuer à la sous-dominance des hybrides. Pour rappel, l'épigénétique fait généralement référence aux éléments héréditaires qui affectent le phénotype de la progéniture sans affecter la séquence d'ADN de la progéniture. Lorsqu'un allèle a été modifié de manière épigénétique, il est appelé
il s'agit d'un épiallèle silencieux. Son paralogue HISN6A est donc essentiel. Chez les hybrides, quand l’épiallèle HISN6B est transmis avec une version mutée de HISN6A, l’hybride est non viable. Cette observation est en accord avec le modèle de Lynch et Force car l'épiallèle héréditaire, qui ne pose généralement pas de problème pour les populations non hybrides, devient problématique lorsqu'il s'agit de la seule copie du gène dans la population hybride (Blevins, Wang, Pflieger, Pontvianne, & Pikaard, 2017).
Les bases génétiques de l’hétérosis
La sélection des hybrides (plantes ou animaux) pour leur phénotype supérieur commença bien avant la compréhension des bases génétiques de cette différence. Au début du XXe
siècle, après la compréhension et l’acceptation des lois de Mendel, les généticiens ont commencé à investiguer les bases génétiques de l’hétérosis des hybrides. Deux hypothèses s’affrontaient alors.
D’une part, l’hypothèse de dominance a été proposée en 1908 par le généticien Charles Davenport (Davenport, 1908). Cette hypothèse attribue la supériorité des hybrides à la suppression des allèles indésirables récessifs d’un parent par des allèles dominants de l’autre parent. Ainsi, l’hétérosis serait directement proportionnelle au nombre d’allèles dominants chez les parents. D’autre part, l’hypothèse de la sur-dominance a été développée de façon indépendante par Edward M. East (1908) et George Shull (1908) (East, 1908; Shull, 1908). Cette hypothèse postule que certaines combinaisons d’allèles obtenues par croisement de deux espèces sont avantageuses chez l’hétérozygote. L'hypothèse de la sur-dominance attribue l’hétérosis à un avantage des génotypes hétérozygotes par rapport aux génotypes homozygotes à un même locus. Cette hypothèse est couramment invoquée pour expliquer la persistance de certains allèles nocifs pour les homozygotes. Dans des circonstances normales, de tels allèles nuisibles seraient éliminés d'une population par le processus de sélection naturelle. Finalement, l’hétérosis pourrait aussi être liée à des mécanismes d’épistasie, par exemple lorsque l’interaction entre deux (ou plusieurs) allèles favorables issus de parents différents conduit à un phénotype amélioré. Les marqueurs moléculaires et les cartes de liaison génétique ont grandement facilité l’identification des loci impliqués dans l’hétérosis. Des études d’analyses QTL (« Quantitative Trait Loci ») ont notamment été conduites chez le riz, démontrant que l’hétérosis résulte en fait d’une combinaison de mécanismes de dominance, sur-dominance et épistatiques (Li et al., 2008).
Tout comme c’était le cas pour la sous-dominance, des mécanismes épigénétiques semblent aussi avoir un rôle dans la mise en place de l’hétérosis (Baranwal, Mikkilineni, Zehr, Tyagi, & Kapoor, 2012; Han & Sapienza, 2008). Il a été démontré que l’hétérosis, dans un hybride de deux espèces d'Arabidopsis, était attribuable à un contrôle épigénétique dans les régions en amont de deux gènes. Le mécanisme implique l'acétylation et /ou la méthylation d'acides aminés spécifiques dans l'histone H3, une protéine étroitement associée à l'ADN, qui peut activer ou réprimer des gènes liés à l’accumulation de chlorophylle et d’amidon. Grâce à cette régulation épigénétique, les hybrides ont une meilleure croissance et une plus grande biomasse par rapport aux parents (Zhongfu et al., 2008).
L’ensemble de ces études nous mène à la constatation que les hybrides ont un génotype et un phénotype uniques. Pourtant, peu d’études s’intéressent aux liens qui existe entre les deux (Xing, Sun, & Ni, 2016). Par ailleurs, ces liens peuvent être multiples et combinés (Figure I) et les études qui s’y intéressent, en utilisant un grand nombre de données, laissent parfois des explications confuses sur les mécanismes dominants (Vacher & Small, 2019). Des modifications du génome, de l’épigénome, du transcriptome, du métabolome, du protéome, du RNAome et des modifications post-traductionnelles ont déjà été observées chez les hybrides (Gang et al., 2009; G. Michael et al., 2011; Mohayeji et al., 2014; Vacher & Small, 2019; Wang, Xue, & Wang, 2014; Wen et al., 2015). Malgré cette complexité apparente, les protéines semblent être un élément clé dans la détermination du phénotype des hybrides. Par exemple, à l’échelle de l’expression des gènes, il a été suggéré que les interactions entre deux génomes parentaux mènent à la modification des transcrits et à une modification de l’abondance des protéines chez les hybrides (Guo et al., 2014; Song et al., 2007). Ainsi, même si les protéines ne sont qu’un seul des éléments expliquant les phénotypes différents des hybrides, leur étude constitue la pièce centrale dans l’assemblage complet du puzzle.
Le lien manquant entre génotype et phénotype : réseaux d’interactions protéiques
Comme mentionné précédemment, les réseaux protéiques jouent un rôle central dans la connexion entre le génotype et le phénotype. Cependant, très peu d’études se sont intéressées au devenir du réseau d’interactions après hybridation. Chez les hybrides, on s’attend à ce que les PIN des deux espèces soient complètement « mélangés » (Diss et
al., 2013) (Figure II). L’interactome qui en résulte chez l’hybride est composé d’interactions intra-espèces (interactions parentales) et d’interactions inter-espèces (interactions chimériques). La perturbation de l’interactome chez l’hybride peut résulter soit en une perte d’interactions entre protéines qui interagissent normalement chez les espèces parentales, soit en la formation de nouvelles interactions spécifiques à l’hybride. L’apparition de nouvelles interactions pourrait résulter des cas où la sélection négative empêche l’interaction des protéines au sein de chaque espèce parentale, mais chez les hybrides cette sélection négative n’est pas maintenue puisque les protéines des espèces parentales n’ont pas co-évolué pour s’éviter. Au contraire, la perte d’interactions pourrait être liée à une incompatibilité entre protéines provenant de génomes différents ; la sélection naturelle favorise le maintien des PPi au fil du temps, établissant la co-évolution de protéines qui interagissent entre elles et empêchant les interactions délétères. Cette co-évolution pourrait induire la perte des PPI ou la formation d’interactions indésirables lorsque deux protéomes différents co-existent dans la même cellule (Lovell & Robertson, 2010).
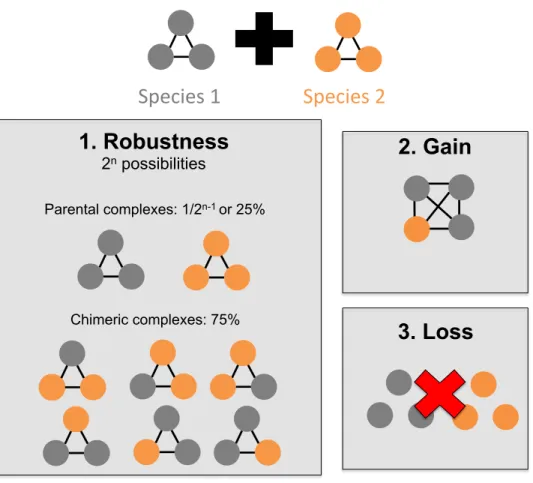
Quelques travaux théoriques se sont intéressés à comprendre l’évolution des PIN après hybridation. Par exemple, Livingston et al. suggèrent que des mutations qui surviennent de façon indépendante chez les espèces parentales pourraient résulter en la formation d’interactions protéiques délétères lorsque combinées chez les hybrides (Livingstone et al., 2012). Il s’agit ici d’un modèle similaire à celui de Bateson-Dobzhansky-Muller, mais appliqué dans le contexte des interactions protéiques. Ce modèle propose aussi que les perturbations du réseau puissent être compensées par la haute connectivité de celui-ci. De façon similaire, un nombre relativement faible d’études ont utilisé des outils expérimentaux pour comprendre les bases moléculaires de l’hétérosis et de la sous-dominance à l’échelle de l’interactome. Lyad et al. ont utilisé des espèces très divergentes de levures pour étudier le réseau d’interaction de la protéine PCNA (Proliferating Cell Nuclear Antigen). Ils ont démontré que la divergence entre espèces parentales peut perturber les PPI chez les hybrides ; la co-évolution des protéines parentales appartenant au réseau d’interactions de la protéine PCNA entraîne la perte des interactions chez les hybrides (Lyad et al., 2012). La même année, Leducq et al. ainsi que Piatkowska et al. publient dans deux études distinctes des résultats contraires à ceux de Lyad et al.. Ces deux études démontrent non pas une perturbation, mais une robustesse des complexes protéiques après hybridation (Leducq et al., 2012; Piatkowska, Naseeb, Knight, & Delneri,
2013). Dans leur étude, Leducq et al. ont généré des hybrides entre deux espèces de levures (Saccharomyces cerevisiae et Saccharomyces kudriavzevii) et ont étudié chez les hybrides l’architecture de deux complexes protéiques : l’ARN polymérase II et le complexe du pore nucléaire. Ils ont démontré que les PPI étaient très conservées chez les hybrides par rapport aux espèces parentales, suggérant que ces complexes ne seraient pas le contributeur majeur de la sous-dominance observée chez les hybrides. Piatkowska et al. ont quant à eux étudié des hybrides entre S. cerevisiae et S. mikatae ou S. uvarum. Après l’étude de six complexes protéiques, ils ont abouti à une conclusion similaire avec un maintien des complexes chez les hybrides. Les deux études ont aussi mis en évidence la présence d’interactions chimériques dans les complexes des hybrides. Cette mise en place d’interactions chimériques chez les hybrides pourrait favoriser non pas la sous-dominance des hybrides, mais l’innovation phénotypique et l’hétérosis. Par exemple, Piatkowska et al. ont démontré que, dans le cas du complexe TRP2/TRP3, la formation d’un complexe chimérique chez les hybrides résulte en un avantage de fitness chez les hybrides dans un environnement sans tryptophane.
À la lumière de ces observations, il apparaît que les complexes protéiques peuvent suivre différentes voies d’évolution après hybridation (figure 1). Les résultats obtenus dans les études précédentes sont assez contrastés, certaines suggèrant une robustesse des complexes (Leducq et al., 2012; Piatkowska et al., 2013), d’autres concluant plutôt une perturbation du réseau d’interactions après hybridation (Lyad et al., 2012). Une limitation que l’on peut noter pour l’ensemble de ces études est qu’elles s’intéressent à un nombre limité de complexes dans la cellule. Lyad et al. ont étudié un réseau d’interactions spécifique (celui de la protéine PCNA), Leducq et al. se sont intéressés à deux complexes protéiques (le complexe du pore nucléaire et l’ARN polymérase II) et Piatkowska et al. ont étudié six complexes (des petits complexes essentiels dont le nombre de sous-unités variait entre deux et quatre). Le défi qui reste à relever est d’explorer le devenir des réseaux d’interactions après hybridation à l’échelle de l’interactome entier. Étant donné le nombre très élevé d’interactions protéiques dans la cellule et les possibilités de perturbations génétiques infinies, il y a nécessité de développer de nouveaux outils expérimentaux et computationnels qui aideront à avoir une vision plus globale du devenir des réseaux d’interactions après hybridation.
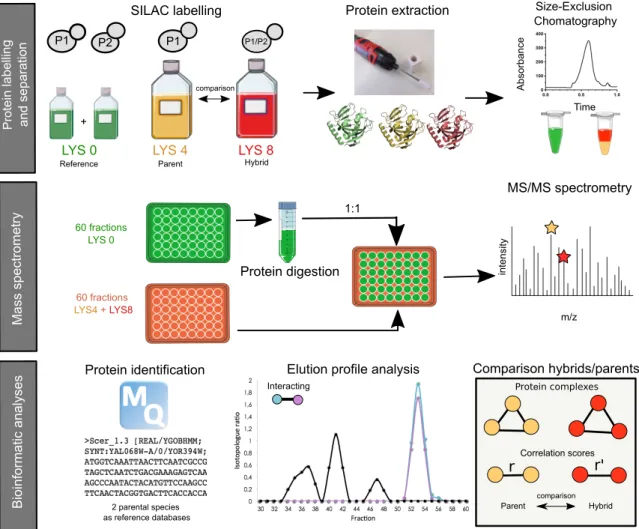
Méthodes d’étude des réseaux d’interactions protéiques
À l’heure actuelle, il existe deux grands types de méthodes qui permettent d’étudier la composition d’un réseau d’interactions protéiques. Le premier type de méthode permet de déterminer les interactions physiques entre protéines par complémentation fonctionnelle (1.6.1). Les autres méthodes permettent quant à elles de déterminer la composition des complexes protéiques par spectrométrie de masse (1.6.2).
Détection des interactions physiques entre protéines par complémentation
fonctionnelle
Ces méthodes reposent sur la reconstitution d’un rapporteur fonctionnel qui émet un signal lorsque les deux protéines s’associent. Les deux protéines à tester sont chacune fusionnées à un domaine différent d’une même protéine, et exprimées ensemble dans une cellule rapporteuse. L’interaction entre les deux protéines recombinantes est détectée in vivo dans la cellule rapporteuse, car elle permet la reconstitution d’une activité biologique. L’activité biochimique restaurée peut être une activation de la transcription (Y2H Yeast Two-Hybrid), une activité enzymatique (PCA Protein-fragment Complementation Assay), ou la fluorescence d’une protéine (FRET Fluorescence Resonance Energy Transfer). La technique de double-hybride (Y2H) a été développée en 1989 par Stanley Fields pour détecter des interactions protéine-protéine dans la levure S. cerevisiae (Stanley & Ok-Kyu, 1989). Le Y2H est basé sur le fait que l’activateur de transcription Gal4p de la levure S. cerevisiae est composé de deux domaines indépendants d’activation de la transcription et de liaison à l’ADN. La protéine appât dont on veut connaitre les interactants est fusionnée au domaine de liaison à l’ADN du facteur de transcription. Les protéines proies sont quant à elles fusionnées au domaine d’activation de la transcription du facteur de transcription. Lorsque la protéine proie est capable d’interagir avec la protéine appât, le domaine d’activation se retrouve à proximité d’un gène rapporteur et la transcription a lieu. L’expression du gène rapporteur permet soit la croissance de la levure sur un milieu auxotrophe de sélection spécifique, soit l’expression d’enzymes. Cette méthode présente toutefois plusieurs limitations. Elle se limite tout d’abord à l’étude de protéines solubles, bien que des variations de la méthode aient été apportées pour l’étude de protéines membranaires (Saraon et al., 2017; Snider, Kittanakom, Curak, & Stagljar, 2010). Ensuite, puisqu’elle implique un facteur de transcription, les interactions testées doivent être localisées dans le noyau, modifiant la localisation endogène des protéines. Cette
technique est peu sensible, non quantitative et s’accompagne de bruit de fond. Cependant, malgré ces contraintes, l’efficacité et le faible coût d’exécution de ce système a permis l’étude des interactions protéiques dans de nombreux organismes. La méthode est aujourd’hui utilisée dans de nombreux systèmes tels que les cellules de mammifères, plantes, insectes ou étoiles de mer (Choi et al., 2003; Ehlert et al., 2006; Luo, Batalao, Zhou, & Zhu, 1997; Mon et al., 2009).
La PCA est une méthode très similaire mais elle se distingue du Y2H par le fait qu’elle utilise non pas un facteur de transcription comme rapporteur, mais une protéine clivée en deux fragments. Cette méthode a notamment été développée chez la levure en utilisant comme rapporteur une version mutée de l’enzyme dihydrofolate réductase (DHFR), conférant à la cellule une résistance au méthotrexate (MTX). Cette enzyme est essentielle à la croissance cellulaire et intervient notamment dans les réactions de synthèse de certaines bases de l’ADN (les purines et la thymine). Lors du test PCA, l’enzyme DHFR est divisée en deux fragments qui ne peuvent pas fonctionner de manière isolée et les deux protéines dont on veut tester l’interaction sont couplées à ces fragments. Lorsque les deux protéines s’associent, la DHFR est reconstituée et la levure est capable de croître sur MTX (Stephen W. Michnick, Ear, Landry, Malleshaiah, & Messier, 2010; Tarassov et al., 2008). Cette méthode présente trois avantages majeurs par rapport au Y2H. Tout d’abord, elle est quantitative. Le signal observé correspond à la densité des cellules (i.e. le nombre de cellules capables de croître sur le milieu de sélection MTX) (Freschi, Torres-Quiroz, Dubé, & Landry, 2013). Ensuite, la localisation des protéines semble conservée (Tarassov et al., 2008), mais il faut toutefois garder en tête que les fragments rapporteurs ajoutés du côté C-terminal des protéines pourraient interférer avec leur signal de localisation, leur repliement et leurs fonctions (Rochette et al., 2015). Finalement, cette méthode réduit de façon importante le bruit de fond par comparaison avec le Y2H. Dans le cas de la PCA, les interactions entre protéines sont visualisées directement, et non pas au travers d’évènements secondaires comme c’est le cas avec le Y2H qui repose sur une activation transcriptionnelle (S. W. Michnick, 2001).
On peut citer comme autres techniques la co-immunoprécipitation ou encore la méthode FRET (Fluorescence Resonance Energy Transfer). La co-immunoprécipitation utilise un anticorps contre une protéine X, sur laquelle une protéine Y vient normalement se coller. L’anticorps qui a reconnu X est immobilisé par une bille de protéine A sépharose
permettant la capture du complexe bille-anticorps X-Y. Les différents acteurs de l’interaction sont ensuite identifiés par transfert de protéines (Western Blot) (Brand, 2006; J.-S. Lin & Lai, 2017). La méthode FRET détecte quant à elle la proximité de molécules fluorescentes (Anca et al., 2016; Day, 2014). La première protéine (le « donneur ») est excitée par une longueur d’onde λ1 et va libérer des photons de longueur d’onde λ2 que l’on peut mesurer. Une partie de l’énergie d’excitation est transférée à la seconde molécule fluorescente (le « receveur ») qui libèrera aussi des photons à une longueur d’onde λ3. Le transfert d’énergie du donneur au receveur, détecté par l’émission de lumière λ3, indique que le donneur et le receveur ont été proches l’un de l’autre. Une variante de la méthode FRET utilise des protéines fluorescentes (comme la GFP) comme donneur et accepteur, ce qui permet d’utiliser la méthode dans un contexte in vivo (méthode BRET Bioluminescent Resonance Energy Transfer) (Truong & Ikura, 2001). Le point fort de cette méthode réside dans l’utilisation de la fluorescence qui permet de suivre en microscopie la localisation cellulaire des protéines avec une bonne résolution spatiale. De plus, le fort signal de fluorescence peut permettre de déterminer la nature des protéines en interaction, la dynamique de ces phénomènes et leur localisation cellulaire. Toutefois, la principale limite reste la difficulté de s’affranchir des signaux parasites (bruits de fond).
Notons pour finir que ces méthodes peuvent révéler à la fois des interactions directes et indirectes. Un résultat positif peut indiquer que les deux protéines interagissent directement, ou de façon indirecte via l’action de « ponts moléculaires » incluant protéines, acides nucléiques (ADN ou ARN) et autres molécules.
Détection des complexes protéiques par purification par affinité et spectrométrie de
masse
Ces méthodes reposent sur la purification de complexes protéiques et l’identification de leurs composantes par spectrométrie de masse (AP-MS Affinity Purification and Mass Spectrometry) (Guillaume et al., 1999; Lee, Adamchek, Feke, Nusinow, & Gendron, 2017). La purification par affinité TAP (Tandem Affinity Purification) sépare une protéine d’intérêt et ses interactants d’un extrait protéique en conditions non dénaturantes (Marcilla & Albar, 2013; Woods et al., 2014). La purification se déroule en 3 étapes : (1) La protéine recombinante est fusionnée à un marqueur (tag). Le marqueur est constitué de trois éléments fusionnés qui vont permettre la double purification : un peptide liant la