Syndrome hémiconvulsion-hémiplégie-épilepsie : à propos de 21 cas
Texte intégral
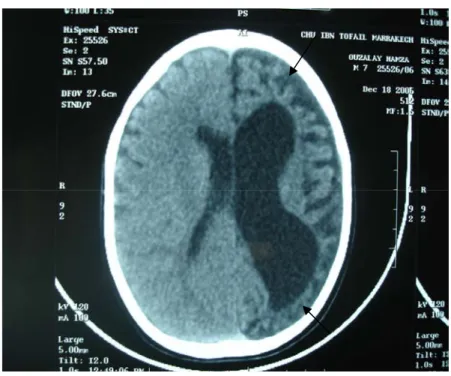
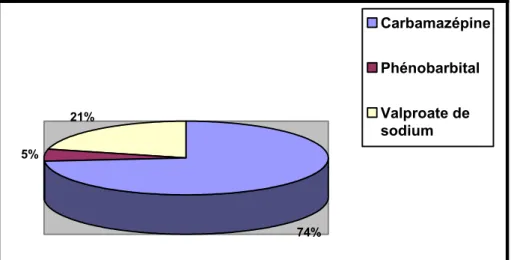
Figure
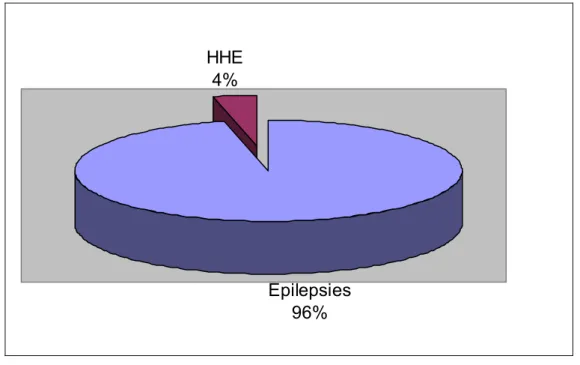
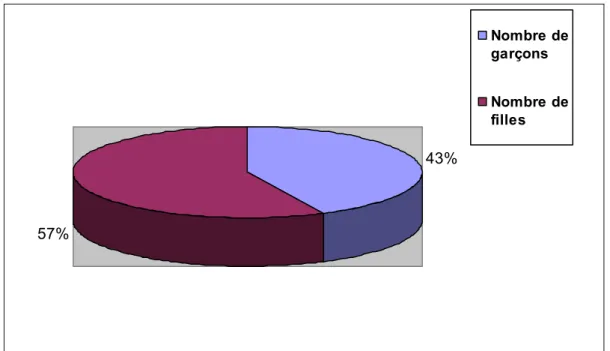
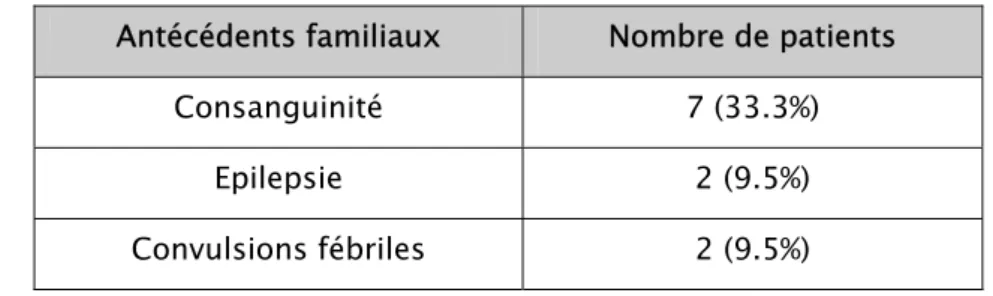
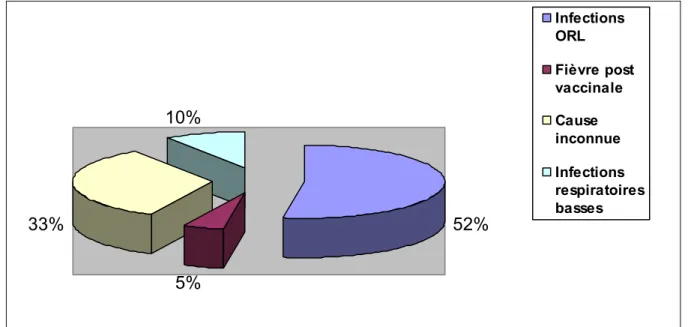
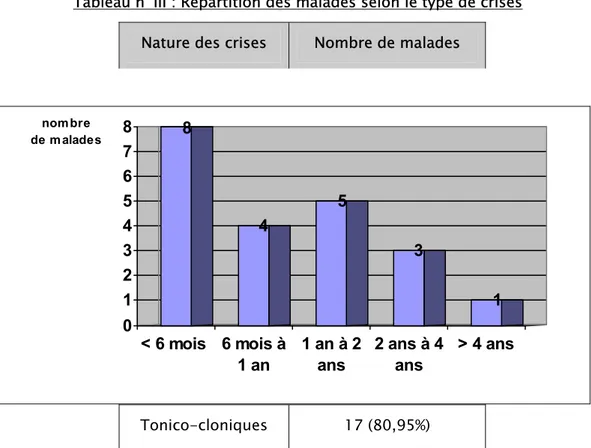
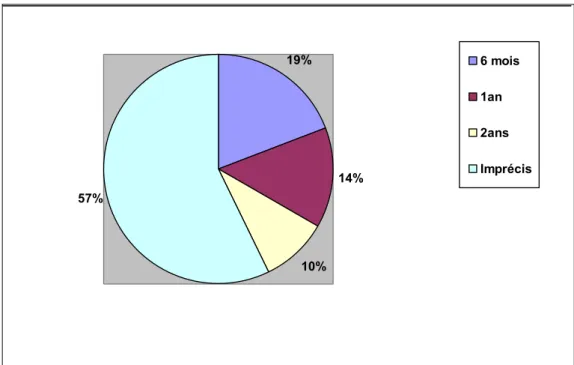
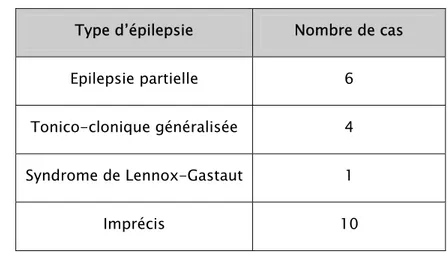
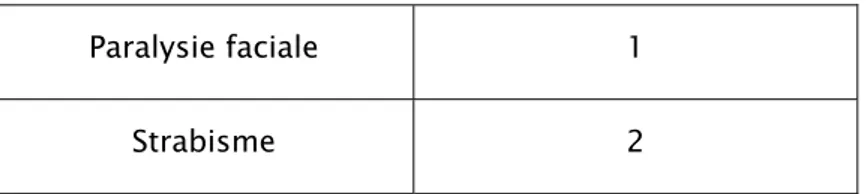
Documents relatifs
* la fièvre a été notée chez 14 malades, soit 23,3٪ des cas, il s’agissait surtout des patients ayant une poussée modérée à sévère de la maladie.. * la tachycardie a
Pour 2 malades, le diagnostic était posé en per-opératoire devant la constatation de bile dans le champ opératoire, pour 15 malades le traumatisme était découvert
Dans notre étude, l’hémoculture a été faite chez tous nos malades, elle était positive dans 65% des cas.. Une infection de la voie urinaire ou rénale peut être le point de départ
En absence d’autre classification, celle de Weiss modi- fiée par Bisceglia était à utiliser pour classifier les TCCO et prédire leur comportement.. La prise en charge de ces
Concernant la prévention primaire des Maladies Cardiovasculaires, une prise en charge.. globale était préconisée avec prise en compte des facteurs de risque
En outre, les dispositions retenues par les Pouvoirs publics concernant la prise en charge des indemnités journalières des professionnelles malades ou confinées sont
la prise en compte du tableau clinique révélateur (fièvre, syndrome inflammatoire biologique, hémocultures, ATCD) du contexte de survenue (drogué IV , infection pulmonaire
Mots-clés : Testicule féminisant- syndrome d’insensibilité aux androgènes- diagnostic- prise en charge thérapeutique et psychologique- Statut juridique. Le syndrome
