© Claire Dziengelewski, 2018
Mécanismes régulateurs de la dynamique de la protéine
E4orf4 de l'adénovirus et de son activité tumoricide
-Implication dans le remodelage de l'enveloppe nucléaire
Thèse
Claire Dziengelewski
Doctorat en biologie cellulaire et moléculaire
Philosophiæ doctor (Ph. D.)
ii
Mécanismes régulateurs de la dynamique de la protéine
E4orf4 de l’adénovirus et de son activité tumoricide
Implication dans le remodelage de l’enveloppe nucléaire
Thèse
Claire Dziengelewski
Sous la direction de :
iii
Résumé
La protéine E4orf4 de l’adénovirus humain est membre d’une famille de facteurs tumoricides, qui induisent la mort cellulaire des cellules cancéreuses tout en épargnant les cellules normales. Il a été montré que dans les cellules transformées, E4orf4 induit un programme de mort cellulaire non apoptotique dépendante des kinases Src et d’une dérégulation de la dynamique d’actomyosine. Cette activité toxique d’E4orf4 est déterminée notamment par sa localisation intracellulaire : E4orf4 est initialement nucléaire, puis elle s’accumule graduellement dans le cytoplasme où elle initie un remodelage atypique du cytosquelette d’actine associé à la mort cellulaire. L’inhibition de l’accumulation cytoplasmique d’E4orf4 empêche également le remodelage de l’actine, et vice versa. Il existe donc un lien fonctionnel entre la dynamique subcellulaire d’E4orf4, le remodelage du réseau d’actine, et l’activité toxique d’E4orf4 ; toutefois, les mécanismes moléculaires impliqués demeurent peu compris. Les travaux présentés dans cette thèse avaient pour but d’identifier les effecteurs cellulaires contrôlant la localisation d’E4orf4 et d’étudier leur implication dans son activité tumoricide.
Des analyses protéomiques ont identifié la protéine de polarité Par3 (PARD3) comme un partenaire d’E4orf4 requis pour l’induction de son activité toxique dépendante de l’actine. Les résultats suggèrent que Par3 et ses partenaires formant le complexe de polarité Par sont impliqués dans le remodelage d’un réseau périnucléaire d’actomyosine dans les cellules tumorales. Ce phénomène stimule la formation et la rupture de blebs de noyau, menant à des bris transitoires de l’enveloppe nucléaire et permettant l’accumulation cytoplasmique d’E4orf4. D’autre part, nous avons mis en évidence qu’E4orf4 stimule le recrutement du co-chaperon BAG3 et de la machinerie autophagique aux sites de formation des blebs nucléaires. Les résultats suggèrent qu’une voie autophagique dépendante de BAG3 est induite en réponse à E4orf4 afin de faire face au stress mécanique, laquelle pourrait contribuer à maintenir l’intégrité et/ou à réparer l’enveloppe nucléaire après sa rupture. En outre, Par3 et BAG3 contribuent également au remodelage de l’enveloppe nucléaire induit par une diminution de sa rigidité par déplétion des lamines en absence d’E4orf4. Ces résultats suggèrent qu’ils participent à la régulation normale de la mécanique nucléaire dans les cellules cancéreuses.
iv
Ainsi, les travaux présentés dans cette thèse appuient un rôle inconnu jusqu’alors pour Par3 et BAG3 dans la plasticité de l’enveloppe nucléaire, qui émerge comme un processus important de la biologie des cellules cancéreuses, contribuant notamment à la migration et à l’invasion tumorale. Cette voie de remodelage du noyau impliquant Par3 et BAG3 pourrait offrir de nouvelles opportunités pour le développement de thérapies moléculaires ciblées.
v
Abstract
The human adenovirus E4orf4 protein is considered as a tumoricidal factor, as it induces cell death in tumor cells while exhibiting low toxicity in non-transformed cells. It was demonstrated that E4orf4 engages a non-apoptotic cell death program, which depends on Src-family kinases and perturbations of actomyosin dynamics. This toxic activity relies on E4orf4 localization dynamics: while E4orf4 is initially nuclear, it gradually accumulates in the cytoplasm where it initiates an atypical remodeling of the actin cytoskeleton associated with cell death. Inhibition of E4orf4’s cytoplasmic accumulation also inhibits actin remodeling, and vice versa. Thus, current data support a functional connection between E4orf4’s subcellular dynamics, actin remodeling, and E4orf4-induced cell death in tumor cells; however, the molecular mechanisms involved are misunderstood. The work presented in this thesis was aimed at identifying key cellular effectors that control E4orf4’s localization dynamics and interrogating their role in the regulation of E4orf4’s tumoricidal action.
We identified the polarity protein Par3 (PARD3) as a binding partner of E4orf4 that is required for the induction of its actin-dependent toxic activity, using proteomic, biochemical and cellular approaches. The results suggest that Par3 and the other Par complex proteins are involved in E4orf4-induced remodeling of the perinuclear actin network in tumor cells. The resulting increase in perinuclear contractility stimulates the formation and rupture of nuclear blebs, leading to transient breaks in the nuclear envelope, cytoplasmic accumulation of E4orf4, and loss of nuclear compartmentalization. Moreover, we found that E4orf4 triggers the recruitment of the co-chaperone BAG3 and the autophagic machinery at nuclear blebs formation sites. The results suggest that a BAG3-dependent autophagy pathway is induced in response to E4orf4 to cope with mechanical stress, which could participate in maintaining the integrity and/or repairing the nuclear envelope after its rupture. Furthermore, Par3 and BAG3 also regulate nuclear envelope remodeling and integrity in cells depleted of lamins, suggesting that these E4orf4 targets may be bona fide regulators of nuclear mechanics in cancer cells.
Thus, the results presented in this thesis support a so-far unappreciated role for Par3 and BAG3 in nuclear envelope plasticity, which emerges as a crucial process regulating cancer
vi
cell biology, notably during tumor cell migration and invasion. This nuclear remodeling pathway involving Par3 and BAG3 could offer new opportunities for the development of targeted molecular therapies.
vii
Table des matières
Résumé ... iii
Abstract ... v
Liste des tableaux ... xi
Liste des figures ... xii
Liste des abréviations ... xiv
Remerciements ... xviii
Avant-propos ... xxi
1 Chapitre 1 : Introduction ... 1
1.1 La mort cellulaire programmée ... 1
1.1.1 Historique de la découverte de l’apoptose ... 1
1.1.2 L’apoptose classique ... 3
1.1.3 Mécanismes de mort cellulaire non apoptotiques ... 5
1.1.3.1 L’apoptose non classique... 7
1.1.3.2 La mort cellulaire par autophagie ... 7
1.1.4 Les protéines tumoricides... 9
1.2 La protéine E4orf4 de l’adénovirus ... 11
1.2.1 E4orf4 et la réplication virale ... 11
1.2.2 Action tumoricide d’E4orf4 ... 13
1.2.3 Signalisation de la mort cellulaire induite par E4orf4 ... 15
1.2.3.1 Rôle de Src ... 16
1.2.3.2 Rôle de PP2A ... 21
1.2.4 Modèles in vivo de l’effet d’E4orf4 ... 23
1.2.5 E4orf4 et la réponse aux dommages à l’ADN ... 25
1.2.6 Identification de nouveaux effecteurs d’E4orf4 : analyse de son interactome .. 27
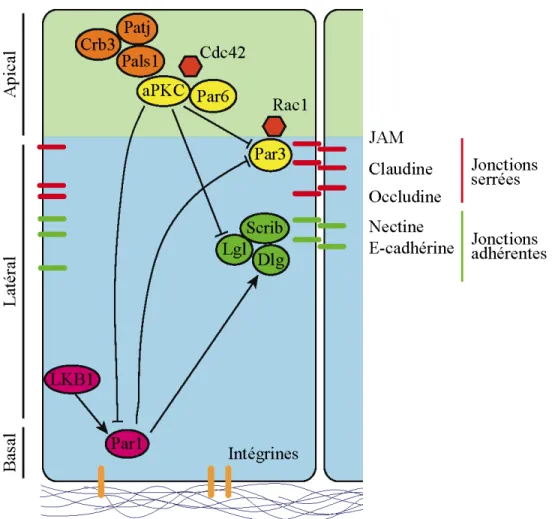
1.3 La protéine Par3 et la polarité apico-basale... 28
1.3.1 Établissement et fonctions de la polarité apico-basale ... 28
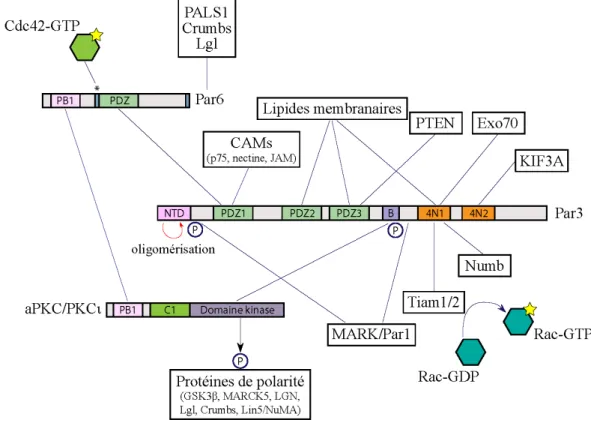
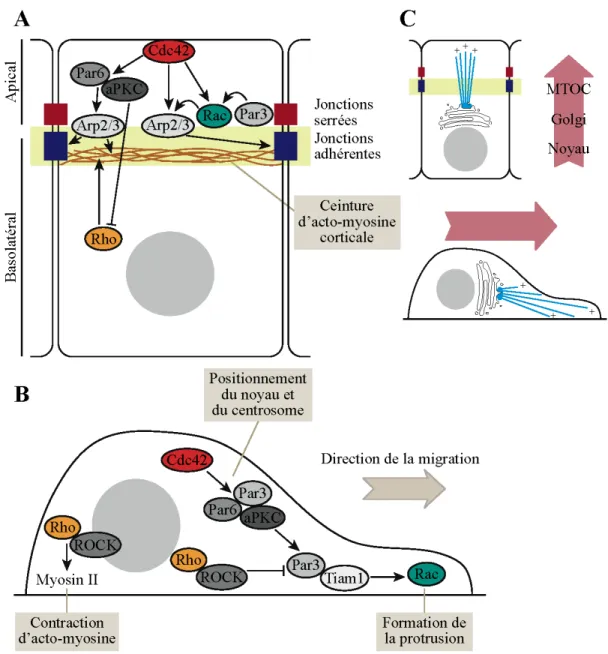
1.3.2 Par3 et le complexe Par ... 31
1.3.2.1 Structure des protéines du complexe Par ... 31
viii
1.3.3 Complexe Par et cancer ... 40
1.3.3.1 Rôles oncogénique et de suppresseur de tumeur des kinases aPKC ... 40
1.3.3.2 Par3 et suppression de la progression tumorale... 42
1.3.3.3 Fonction pro-tumorale de Par3 ... 43
1.3.4 Rôle du complexe Par dans l’export nucléaire par formation de blebs nucléaires 46 1.4 Dynamique de l’enveloppe nucléaire ... 48
1.4.1 Structure de la membrane nucléaire ... 48
1.4.2 LINC et lamina : rôles physiologiques et pathologiques ... 51
1.4.3 Régulation de l’intégrité de l’enveloppe nucléaire en interphase ... 54
1.5 La protéine BAG3 et les mécanismes d’autophagie sélective ... 56
1.5.1 Le contrôle de qualité des protéines ... 56
1.5.1.1 La dégradation par le protéasome ... 57
1.5.1.2 La dégradation par autophagie ... 57
1.5.2 Autophagie et homéostasie nucléaire ... 59
1.5.3 La protéine BAG3 ... 61
1.5.3.1 Structure de BAG3 ... 61
1.5.3.2 Autophagie sélective dépendant de BAG3 ... 62
1.5.3.3 BAG3 et le cancer... 64
1.6 Contexte, hypothèse et objectifs ... 65
2 Chapitre 2 : Targeting of Tumor Cells via Par3 Polarity Protein-Regulated Mechanical Force-Induced Nuclear Envelope Rupture ... 67
2.1 Avant-propos ... 68
2.2 Résumé ... 70
2.3 Abstract ... 71
2.4 Introduction ... 72
2.5 Results ... 74
2.5.1 E4orf4’s tumoricidal activity correlates with changes in perinuclear actomyosin organization that modulate its spatial dynamics. ... 74
2.5.2 E4orf4 interacts with Par complex proteins in a Par3-dependent manner and promotes epithelial cell polarity. ... 76
ix
2.5.3 E4orf4’s interaction with Par3 enables corruption of actomyosin dynamics and
promotes E4orf4 nuclear exit in tumorigenic cells. ... 80
2.5.4 Par3 promotes nuclear exit of E4orf4 via actomyosin-dependent nuclear bleb formation. ... 86
2.5.5 E4orf4 engages a positive feedback loop implicating actomyosin contractility and nuclear envelope rupture, which impairs nuclear compartmentalization. ... 89
2.5.6 Par3 regulates actin-dependent nuclear mechanics... 92
2.6 Discussion ... 94
2.7 Material and methods ... 101
2.8 References ... 110
2.9 Supporting information captions ... 123
3 Chapitre 3 : Modulation de l’activité d’E4orf4 par le co-chaperon BAG3 : mise en évidence d’un rôle pour BAG3 dans le maintien de l’intégrité de l’enveloppe nucléaire ... 125
3.1 BAG3 régule la localisation noyau-cytoplasme d’E4orf4 et son activité toxique ... 127
3.2 E4orf4 induit le recrutement de facteurs autophagiques dans la région périnucléaire de manière dépendante de BAG3 ... 131
3.3 La déplétion de BAG3 affecte la dynamique de formation et rupture des blebs nucléaires induits par E4orf4 ... 139
3.4 BAG3 module la réparation de l’enveloppe nucléaire après des ruptures spontanées induites par un affaiblissement de la lamina ... 143
3.5 La déplétion de BAG3 et l’expression d’E4orf4 favorisent l’apparition d’anomalies nucléaires ... 146
3.6 Modèle et perspectives ... 150
3.7 Matériel et méthodes ... 155
4 Chapitre 4 : Discussion ... 162
4.1 Rôle de la dynamique de l’actine dans l’activité tumoricide d’E4orf4 ... 162
4.1.1 Relation bidirectionnelle entre la dynamique de l’actine et l’activité toxique d’E4orf4 ... 162
4.1.2 Régulation de la fonction de Par3 par la dynamique de l’actine ... 163
4.1.3 La reprogrammation oncogénique de Par3 en tant que déterminant de l’activité tumoricide d’E4orf4 ... 165
x
4.2 Modulation des propriétés mécaniques du noyau par E4orf4 ... 167
4.2.1 Export du noyau par formation et rupture de blebs nucléaires ... 167
4.2.2 Vers l’identification d’un mécanisme d’autophagie sélective pour la dégradation des agrégats protéiques nucléaires ... 168
4.2.3 Régulation mécanique de la forme du noyau dans les cellules transformées ... 169
4.2.4 BAG3 et aneuploïdie ... 172
4.3 E4orf4 et le stress mécanique : impact sur l’activité de YAP/TAZ ... 173
4.3.1 Régulation de YAP par l’expression d’E4orf4 ... 174
4.3.2 Régulation de YAP/TAZ par le complexe Par ... 177
4.3.3 Régulation de YAP par BAG3 ... 178
4.3.4 Régulation de YAP par Src ... 179
4.4 Conclusion ... 180
5 Bibliographie ... 183
6 Annexe 1 ... 215
xi
Liste des tableaux
Chapitre 1
Tableau 1.1 : Caractéristiques des principales protéines tumoricides connues
Annexe 1
Tableau 6.1 : Protéines identifiées en complexe avec E4orf4(WT)mCherry par spectrométrie de masse lors des expériences de co-immunoprécipitation dans les cellules MDA-MB-231.
xii
Liste des figures
Chapitre 1
Figure 1.1 – Voies de signalisation induisant l’apoptose classique.
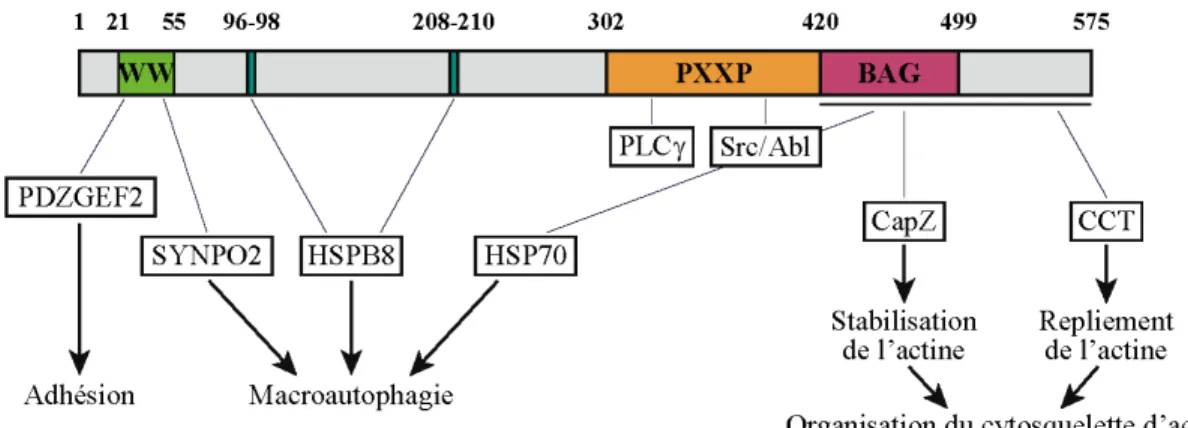
Figure 1.2 – Schéma de la structure de la protéine E4orf4 de l’adénovirus. Figure 1.3 – Déséquilibre de l’activité des Rho GTPases induit par E4orf4.
Figure 1.4 – Organisation des complexes régulant la mise en place et le maintien de la polarité apico-basale des épithéliums.
Figure 1.5 – Schéma des interactions protéiques impliquant les protéines du complexe Par. Figure 1.6 – Schéma du rôle des protéines du complexe Par dans la ségrégation de l’activité des Rho GTPases dans les cellules épithéliales et dans les cellules en migration.
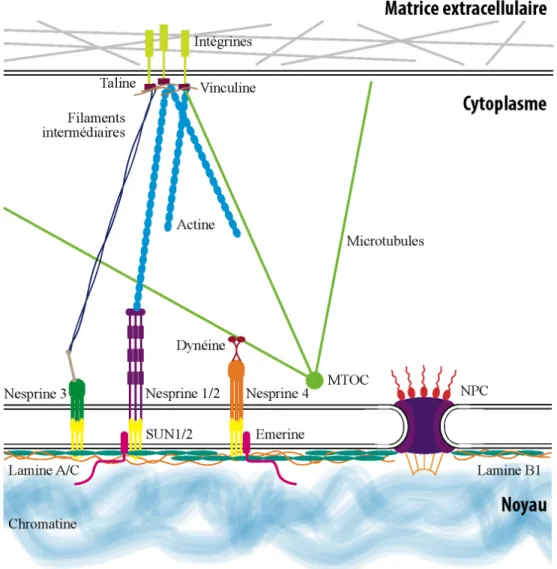
Figure 1.7 – Schéma des complexes protéiques retrouvés à la membrane nucléaire et leur interaction avec le cytosquelette.
Figure 1.8 – Schéma de la voie d’induction de la macroautophagie. Figure 1.9 – Schéma de la structure de la protéine BAG3.
Chapitre 2
Figure 2.1 – E4orf4-induced actin remodeling and spatial dynamics are tumor cell context-dependent.
Figure 2.2 – E4orf4 interacts with Par3 and modulates epithelial cell polarity.
Figure 2.3 – E4orf4-induced cell blebbing and nuclear condensation in tumorigenic cells depends on the scaffolding function of Par3.
Figure 2.4 – Par3 regulates remodeling of the perinuclear actin network and E4orf4 spatial dynamics.
Figure 2.5 – Analysis of the molecular determinants and the subcellular distribution of E4orf4-Par3 interaction.
Figure 2.6 – E4orf4 nuclear exit by nuclear bleb formation is inhibited by silencing of Par3. Figure 2.7 – E4orf4-induced nuclear bleb formation causes NE rupture regulated by the LINC complex.
Figure 2.8 – Spontaneous nuclear envelope rupture due to lamina protein depletion depends on Par3.
Figure S2.1 – Depletion of Par3 inhibits E4orf4-driven membrane blebbing and nuclear condensation in 293T cells.
Figure S2.2 – Par3 binding regulates E4orf4 actin-remodeling activity and localization dynamics.
Figure S2.3 – Controls for the specificity of the Duolink proximity ligation assay to visualize the in situ subcellular interaction of E4orf4 and endogenous Par3.
Figure S2.4 – Characterization of E4orf4-induced nuclear blebbing.
Chapitre 3
Figure 3.1 – BAG3 régule la dynamique noyau-cytoplasme d’E4orf4 et son activité toxique. Figure 3.2 – E4orf4 stimule la machinerie autophagique de manière dépendante de BAG3. Figure 3.3 – La déplétion de BAG3 affecte la dynamique de formation et rupture des blebs nucléaires induits par E4orf4.
Figure 3.4 – BAG3 et l’autophagie modulent la réparation de l’enveloppe nucléaire après une rupture spontanée dans les cellules déplétées en lamines.
xiii
Figure 3.5 – La déplétion de BAG3 combinée à une faible expression d’E4orf4 favorisent l’apparition d’anomalies nucléaires.
Figure 3.6 – Modèle proposé
Figure S3.1 – Caractérisation des lignées cellulaires HeLa TRex RFP et Flag-E4orf4(WT)RFP
Figure S3.2 – Représentation schématique de l’utilisation de LC3B lipidé pour mesurer le flux autophagique.
Chapitre 4
Figure 4.1 – E4orf4 affecte la localisation de YAP de façon dépendante de Par3.
Figure 4.2 – Modèle du lien bidirectionnel entre la dynamique de l’actine et la localisation d’E4orf4 pour son activité toxique dans les cellules transformées.
xiv
Liste des abréviations
3D trois dimensions
53-BP1 TP-53 binding protein 1
ACF ATP-utilizing chromatin assembly and remodeling factor
Ad adénovirus
ADN acide désoxyribonucléique AIF apoptosis-inducing factor
Amotl1/2 angiomotin-like protein 1/2
AMP adénosine monophosphate AMPK AMP-activated protein kinase
Apaf-1 apoptotic protease-activating factor 1
APC/C anaphase-promoting complex/cyclosome
AP-MS affinity purification coupled to mass spectrometry
ARM arginine-rich motif
ARNm acide ribonucléique messager Arp (2/3) actin-related protein (2/3)
ASPP1, 2 apoptosis stimulating proteins of p53
ATG autophagy-related
ATM ataxia telangiectasia mutated
ATP adénosine triphosphate
ATR ataxia telangiectasia and Rad3-related
BAG Bcl-2 associated athanogene
Bak Bcl-2 homologous antagonist killer
Bax Bcl-2-associated X protein
Bcl-2 B-cell lymphoma 2
BID BH3-interacting domain death agonist
BioID biotin identification
BRCA1 breast cancer type 1 susceptibility protein C. elegans Caenorhabditis elegans
C-ter C-terminal Ca2+ ions calcium
CAR Coxsackie virus and adenovirus receptor
Cdc42 cell division control protein 42
CHMP4, 7 charged multivesicular body protein 4
Crb3 Crumbs3
CRIB Cdc42- and Rac-interactive binding domain
Ctl contrôle
dIAP1 death-associated inhibitor of apoptosis 1
Dlg Disks large
E1A early region 1A
E1B 19K early region 1B 19 kilodalton
E1B 55K early region 1B 55 kilodalton
E4orf4, 3, 6 early region 4 open reading frame 4, 3, 6
EGFR epithelial growth factor receptor Egr-1 early growth response protein 1
xv
ErbB2 (avian) erythroblastosis oncogene B2
ERK extracellular signal-regulated kinase
ESCRT endosomal sorting complex required for transport
EV empty vector
FAK focal adhesion kinase
FL full length ; pleine longueur
GAPDH glyceraldéhyde-3-phosphate déshydrogénase GEF guanine exchange factor
GDP guanosine diphosphate GFP green fluorescent protein
GST glutathione S-transférase GTP guanosine triphosphate
HAMLET human alpha-lactalbumin made lethal to tumor cells
HGF hepatocyte growth factor hid head involution defective
HSP heat shock protein
iASPP inhibitor of apoptosis-stimulating protein of p53
IAP inhibitor of apoptosis protein Iex-1 immediate early response gene X-1
IL-10 interleukine-10
INM inner nuclear membrane
IPV isoleucine-proline-valine JAM junctional adhesion molecule
JNK Jun N-terminal kinase
KASH Klarsicht/ANC-1/Syne-1 homology
kb kilobase kDa kiloDalton
KIF3A kinesin family member 3A
LATS1/2 large tumor suppressor kinase 1/2
LC-MS/MS liquid chromatography coupled to tandem mass spectrometry
LC3 (microtubule-associated proteins 1A/1B) light chain 3
Lgl lethal giant larvae
LINC linker of nucleoskeleton and cytoskeleton
LKB1 liver kinase B1
MAPK mitogen-activated protein kinase
MDA-7 melanoma differentiation associated gene 7
MDCK Madin-Darby canine kidney
MLC2 myosin light chain 2
MOI multiplicity of infection
MRCK myotonic dystrophy kinase-related Cdc42-binding kinase
MTOC microtubule organizing center
mTORC1 mechanistic target of rapamycin complex 1
MYPT1 myosin phosphatase target subunit 1
N-ter N-terminal
NBR neighbor of BRCA1
NE nuclear envelope
xvi
NH4Cl chlorure d’ammonium
NLS nuclear localization sequence
NPC nuclear pore complex
NS1 non-structural protein 1
N-WASP neural Wiskott-Aldrich syndrome protein
Omi/HtrA2 Omi/high temperature requirement protein A2
ONM outer nuclear membrane
Pals1 protein associated with lin-7
Patj Pals1-associated tight junction protein
PB1 Phox and Bem1
PBM PDZ-binding motif
PCR polymerase chain reaction
PDK1 phosphoinositide-dependent kinase 1
PDZ PSD95/Dlg1/ZO-1 Phase G2/M phase Gap 2/Mitose
Phase S phase de synthèse de l’ADN PI3K phosphoinositide 3-kinase
PIP(3) phosphatidylinositol (3)-phosphate
(a)PKC (atypical) protein kinase C
PKC-BD protein kinase C binding domain
PLA Proximity Ligation Assay
PP1 protéine phosphatase 1 PP2A protéine phosphatase 2A
PTEN phosphatase and tensin homolog
PTPN14 tyrosine-protein phosphatase non-receptor type 14
PXXP domaine riche en prolines Rab11a Ras-related protein Rab11a
Rac1 Ras-related C3 botulinum toxin substrate 1
RASSF7, 8 Ras association domain-containing protein 7
RDA réponse aux dommages à l’ADN Région E Région Early (gènes précoces) Région L Région Late (gènes tardifs)
(m)RFP (monomeric) Red Fluorescent Protein
RNP ribonucléoprotéines
ROCK Rho-associated protein kinase
rpr reaper
RSQS arginine-sérine-glutamate-sérine
RT room temperature ; temperature ambiante S. cerevisiae Saccharomyces cerevisiae
SCQP système de contrôle de qualité des protéines Scrib Scribble
SE standard error ; erreur standard
SFK Src-family kinases
SH1, 2, 3 Src homology 1, 2, 3
siARN small interfering acide ribonucléique
Smac/DIABLO second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI
xvii
SQSTM1 sequestosome 1
SUN Sad1p/UNC-84
TGFβ transformed growth factor β
Tiam1/2 T-lymphoma invasion and metastasis inducing protein 1/2
TNF tumor necrosis factor
TRAIL TNF-related apoptosis-inducing ligand tws/aar twins/abnormal anaphase resolution
ULK1 UNC-51-like kinase 1
VMP1 vacuole membrane protein 1
VPS vacuolar protein sorting
WT wild-type
YAP/TAZ Yes-associated protein/transcriptional coactivator with PDZ-binding motif
ZO-1 zonula occludens protein 1
xviii
Remerciements
Les travaux présentés dans cette thèse représentent l’essentiel du projet de recherche que j’ai réalisé au cours de ma maîtrise, puis de mon doctorat dans le laboratoire du Dr Josée N. Lavoie. De nombreuses personnes m’ont permis de tirer le meilleur, aussi bien sur le plan académique que personnel, de ces plus de six ans, et ont ainsi contribué à la réussite de mon doctorat.
En premier lieu, je tiens à remercier Josée Lavoie de m’avoir accueillie comme étudiante à la maîtrise après une brève discussion dans son bureau, quelques semaines à peine avant le début de la session d’automne. Merci Josée pour la confiance que tu m’as rapidement accordée. Merci de m’avoir donné l’opportunité de former des étudiants stagiaires, et d’avoir considéré mon avis lors de discussions sur mon projet ou sur d’autres projets du laboratoire. Tu m’as donné la possibilité de profiter de toutes les occasions qui ont pu se présenter pendant mon doctorat, et c’est une réelle chance qui m’a beaucoup appris sur le plan scientifique et personnel. Merci aussi de m’avoir permis de présenter mes travaux en congrès local et international. J’ai beaucoup apprécié me sentir partie intégrante du laboratoire pendant toutes ces années.
Je remercie également Herman Lambert et Margit Fuchs, du fond du cœur. Herman, pour sa bonne humeur éternelle et sa disponibilité légendaire pour résoudre les problèmes de biochimie et les clonages récalcitrants. Merci pour les chocolats et les bonbons que tu t’assures toujours de réapprovisionner. Merci aussi à Giti pour sa bienveillance et sa rigueur scientifique, et pour m’avoir souvent apporté une petite portion de sa bonne recette de la fin de semaine. Merci sincèrement à vous deux pour l’atmosphère sans nuage du laboratoire pendant toutes ces années. On a vraiment beaucoup de chance de travailler dans un environnement aussi agréable, et c’est beaucoup grâce à vous.
J’aimerais remercier les autres membres du laboratoires et étudiants actuels ou passés. Merci à Claudia qui m’a installée dans le laboratoire à mon arrivée et qui m’a tout appris! Merci aussi à Marie-Chloé, Marilène, Alexandra et Émilie. Merci à Marc-Antoine « Ça Se Finit Là » Rodrigue, qui a été mon stagiaire puis mon collègue ; ta rigueur innée en tant que
xix
stagiaire a rendu mon travail de formation extrêmement plaisant et facile. Merci aussi de m’avoir acceptée dans ton équipe au beer-pong à plusieurs reprises malgré mes compétences approximatives.
Je dois aussi remercier toutes les personnes du centre de recherche qui ont contribué de près ou de loin à la réussite de mon projet. Merci à Carl Saint-Pierre pour son aide avec la microscopie et pour son dévouement au fonctionnement des expériences de live cell, qui ne se soucie pas de l’heure du jour ou de la nuit. Merci aussi au Dr. Patrick Laprise et à Clémence Gamblin pour leur implication dans l’initiation d’un projet avec la drosophile. Il n’aura malheureusement pas abouti mais j’ai beaucoup aimé apprendre à travailler avec ce modèle.
Je dois également remercier tous les amis que j’ai rencontrés pendant mes années de maîtrise et de doctorat. Je ne pense pas que j’aurai à nouveau la chance d’avoir un grand groupe d’amis aussi bienveillants les uns envers les autres. Merci à Manu, Alex, Gaëlle, Imène, Lauriane, Alice V., Alice B., Carole, J-C, Cornélia, Myriam, Françoise, Aksam, Khalid, Laurent, Maëva, Kévin, Solenn, Mélany, Mathieu… et tous les autres. Vous m’avez apporté un équilibre sans lequel l’expérience du doctorat aurait été plus ardue. Merci pour tout, merci pour les soirées, les anniversaires, les sorties à Valcartier ou au festival d’été, pour les kilos de poutine club, le billard du dimanche soir et l’occasionnel thé vert du lundi matin. Je remercie aussi Clémence, Sébastien, Manon, Fanny et tous les autres pour les soirées qui font du bien. Merci également à Marilyne pour son amitié attentionnée. Mention spéciale à Carole qui a été ma colocataire pendant tout ce temps et qui confirme que parfois les décisions prises sur un coup de tête fonctionnent le mieux! Je vous remercie tous de m’avoir fait sentir bien entourée pendant toutes ces années. Si c’était à refaire, je ne changerais rien.
Enfin, je tiens à remercier ma famille pour leur soutien et leur présence. Merci à mes parents, Agnès et Hervé, de m’avoir permis de partir au Québec à juste 18 ans (!) et de m’avoir toujours laissée faire mes choix en s’assurant que je ne manque de rien. Je réalise que j’ai beaucoup de chance. Merci à mes sœurs Sophie et Mathilde pour leurs messages réguliers et les bons moments passés en vacances. Je remercie aussi les membres de ma famille qui sont
xx
venus me rendre visite à Québec et qui m’ont permis de découvrir avec eux les paysages québecois.
Je tiens également à remercier mes professeurs de SVT, Mr Cousty et Mme Lesne, qui m’ont transmis leur amour de la biologie. Merci particulièrement à Mme Lesne de m’avoir appris à aimer écrire la science.
Enfin, je remercie les membres de mon jury d’avoir accepté de corriger ma thèse. J’espère que vous en apprécierez la lecture.
xxi
Avant-propos
Les travaux présentés dans cette thèse sont le résultat de mes études de doctorat dans le laboratoire du Dr Josée N. Lavoie, amorcés pendant ma formation à la maîtrise. Au cours de mon doctorat, j’ai participé à la rédaction d’un article en tant que première auteure, présenté en chapitre 2 de cette thèse. Les expériences réalisées contribueront également à un deuxième article en préparation dont les résultats sont présentés au chapitre 3.
L’objectif général de mon doctorat est d’identifier des protéines cellulaires impliquées dans la régulation de l’activité tumoricide de la protéine E4orf4 de l’adénovirus humain.
Dans un premier temps, j’ai identifié la protéine de polarité Par3 comme partenaire d’interaction d’E4orf4 par des analyses protéomiques. J’ai découvert que Par3 contribue à un mécanisme de remodelage nucléaire par la tension, responsable de la régulation de la forme et de l’intégrité du noyau. Ces travaux sont présentés en chapitre 2 de cette thèse et font l’objet d’un article actuellement en révisions dans Journal of Cell Biology, intitulé : « Targeting of Tumor Cells via Par3 Polarity Protein-Regulated Mechanical Force-Induced Nuclear Envelope Rupture ». Les co-auteurs de cet article sont Marie-Chloé Boulanger, Margit Fuchs, Herman Lambert et Josée N. Lavoie.
En complément de ce chapitre, l’annexe 1 présente une liste plus complète des protéines identifiées par spectrométrie de masse comme partenaires d’interaction d’E4orf4.
Les résultats présentés au chapitre 2 ont mis en lumière la pertinence d’utiliser E4orf4 comme un outil moléculaire pour étudier les mécanismes responsables du maintien de l’intégrité et la réparation de l’enveloppe nucléaire dans les cellules transformées. Utilisant E4orf4 comme modèle de remodelage de l’enveloppe nucléaire par le stress mécanique, j’ai identifié un nouveau rôle pour le co-chaperon moléculaire BAG3 et les mécanismes autophagiques qui en dépendent, qui semble contribuer à la réparation de l’enveloppe nucléaire après sa rupture. Ces travaux font l’objet du chapitre 3 de cette thèse.
xxii
A mon arrivée au laboratoire, j’ai également contribué à un article montrant qu’E4orf4 altère la dynamique des endosomes Rab11a et des mitochondries pour mener à la mort cellulaire. L’article issu de ces travaux, présenté en annexe 2, s’intitule : « A Functional Interplay between the Small GTPase Rab11a and Mitochondria-Shaping Proteins Regulates Mitochondrial Positioning and Polarization of the Actin Cytoskeleton Downstream of Src Family Kinases ». Landry, M.-C., Champagne, C., Boulanger, M.-C., Jetté, A., Fuchs, M., Dziengelewski, C. et Lavoie, J. N. Journal of Biological Chemistry, 2014.
Par souci d’harmonisation avec la littérature actuelle, les protéines seront désignées par leur nom usuel dans cette thèse lorsque ce nom est plus communément utilisé que la nomenclature officielle. En cas de confusion possible, le nom officiel de la protéine sera indiqué entre parenthèses.
L’ensemble de ces travaux n’aurait pas pu voir le jour sans la contribution des co-auteurs cités ci-dessus.
1
1
Chapitre 1 : Introduction
Lors de la transformation tumorale, l’une des caractéristiques communément acquises par les cellules consiste en l’échappement aux signaux induisant la mort par apoptose (Hanahan et Weinberg, 2011). Ce phénomène est problématique d’un point de vue clinique, car il limite l’efficacité des thérapies visant à mener les cellules cancéreuses à la mort en stimulant des voies endogènes. Dans ce contexte, l’étude des protéines tumoricides présente un intérêt particulier ; en effet, ces facteurs provoquent la mort des cellules transformées tout en épargnant la survie des cellules normales (Noteborn, 2009). En particulier, plusieurs protéines tumoricides font intervenir des voies non apoptotiques, c’est-à-dire indépendantes de la voie canonique d’apoptose, dérégulée dans les cellules transformées. C’est le cas de la protéine E4orf4 de l’adénovirus, qui fait l’objet de cette thèse.
Bien que les mécanismes cellulaires menant à la mort induite par E4orf4 aient été caractérisés, peu d’éléments permettent d’expliquer l’absence d’effet toxique dans les cellules non transformées. L’activité tumoricide d’E4orf4 suppose que les voies de signalisation avec lesquelles elle interfère dans les cellules transformées ne lui sont pas accessibles dans les cellules normales. Il est donc primordial d’identifier ces voies de signalisation qui devraient être communes à la majorité des cellules transformées et qui, une fois dérégulées, auraient la capacité d’induire leur mort.
Les travaux présentés dans cette thèse ont identifié les protéines Par3 et BAG3 comme des régulateurs de l’activité tumoricide d’E4orf4. Ces deux protéines semblent être impliquées dans le contrôle de la localisation dynamique d’E4orf4 entre le noyau et le cytoplasme, qui est identifiée comme un processus étroitement lié à son activité tumoricide.
1.1 La mort cellulaire programmée
1.1.1 Historique de la découverte de l’apoptose
La mort cellulaire programmée est un mécanisme physiologique d’une importance majeure lors du développement (Miura, 2011). Elle permet d’éliminer les cellules superflues ou anormales et potentiellement dangereuses, afin d’assurer l’homéostasie de l’organisme. Chez
2
l’humain, entre 10 et 100 milliards de cellules meurent chaque jour et sont remplacées par de nouvelles cellules (Renehan et al., 2001).
La première classification morphologique de la mort cellulaire a été réalisée en 1973 à partir d’observations d’embryons de rats exposés à des produits toxiques (Schweichel et Merker, 1973). À la même période, le terme d’apoptose (venant du grec άπόπτωσισ, « la chute ») fut adopté pour désigner un type de mort cellulaire observé dans plusieurs tissus (Kerr et al., 1972). Malgré leurs origines distinctes, les cellules mortes observées partageaient plusieurs caractéristiques morphologiques, suggérant aux auteurs l’existence d’un mécanisme endogène et conservé de mort cellulaire. Ainsi, l’apoptose fut décrite par opposition à la nécrose, processus de mort cellulaire accidentel et incontrôlé (Wyllie et al., 1980). La dichotomie entre apoptose et nécrose reposait principalement sur des critères morphologiques cellulaires. En effet, l’apoptose était alors caractérisée par la condensation cellulaire et de la chromatine, suivie par la fragmentation de l’ADN et de la cellule en corps apoptotiques éliminés par phagocytose (Kerr et al., 1972). A l’inverse, le processus nécrotique était décrit comme un gonflement de la cellule et un déversement du contenu cellulaire dans le milieu extérieur, menant à une réponse inflammatoire de l’organisme (Majno et Joris, 1995).
La découverte des caspases au début des années 1990 a permis d’élucider les processus moléculaires impliqués dans la mort par apoptose (Yuan et al., 1993 ; Miura et al., 1993). L’activation des caspases est alors devenue le critère standard pour différencier la mort apoptotique de la mort accidentelle. L’identification des cibles des caspases et des effecteurs impliqués dans la régulation de l’apoptose a mis en évidence la grande complexité et la régulation croisée des voies signalétiques mises en jeu. L’étude du rôle de ces voies a également révélé l’existence de modalités de mort cellulaire non apoptotiques, mais contrôlables au niveau cellulaire, ajoutant à la complexité de la régulation de la viabilité cellulaire (Tait et al., 2014).
3 1.1.2 L’apoptose classique
Les premières études systématiques de l’apoptose ont été réalisées chez le nématode C.
elegans, et ont souligné l’importance des processus de mort cellulaire programmée dans le
développement de cet organisme (Sulston et al., 1983). Les travaux suivants de la même équipe ont identifié 14 gènes impliqués à différentes étapes de l’induction de l’apoptose dans ce contexte (Hedgecock et al., 1983 ; Ellis et Horvitz, 1986 ; Ellis et al., 1991 ; Hengartner et al., 1992). Les produits de ces gènes présentent des fonctions pro-apoptotiques ou anti-apoptotiques, illustrant la régulation fine du processus de mort cellulaire mis en œuvre lors du développement. Ces découvertes initiales ont eu un impact majeur sur notre compréhension des mécanismes de survie et de mort cellulaire, et les chercheurs à leur origine ont été récompensés par le prix Nobel de physiologie ou de médecine en 2002.
D’un point de vue phénotypique, l’induction de l’apoptose mène à l’arrondissement et à la perte de volume cellulaires, au bourgeonnement (blebbing) de la membrane plasmique, à la condensation de la chromatine (pyknosis) et la fragmentation du noyau (karyorrhexis) (Kerr et al., 1972). La cellule apoptotique se désagrège en corps apoptotiques qui sont ensuite éliminés par les cellules phagocytaires de l’organisme.
Les voies canoniques de l’apoptose reposent sur l’activation des caspases. Ces enzymes sont des protéases à cystéine retrouvées à l’état basal sous forme de pro-caspases inactives (Cohen, 1997). Une fois activées, elles catalysent le clivage de nombreux substrats protéiques, conduisant à l’inactivation de protéines de survie et à la stimulation des fonctions de mort cellulaire (Figure 1.1) (Lüthi and Martin, 2007). L’apoptose peut être déclenchée par des stimuli extracellulaires, via la liaison de facteurs de mort à leurs récepteurs (voie extrinsèque) (Locksley et al., 2001). L’activation des récepteurs de mort cellulaire conduit à l’assemblage de complexes protéiques stimulant l’activation des caspases. L’apoptose peut également être engendrée par des signaux intracellulaires à la suite de stress, tels que les dommages à l’ADN, le stress oxydatif ou l’absence de certains facteurs de croissance (voie intrinsèque). Cette voie est régulée par les protéines de la famille Bcl-2 (Youle et Strasser, 2008). Elle mène à la perméabilisation de la membrane externe de la mitochondrie, provoquant la relâche de protéines mitochondriales dans le cytoplasme telles que le
4
cytochrome C, Smac/DIABLO ou Omi/HtrA2 (Joza et al., 2001). Ces facteurs stimulent l’activation des caspases effectrices afin d’amplifier le signal de mort, ainsi que la fragmentation du génome (Hill et al., 2004; Enari et al., 1998 ; Li et al., 2001). La perte d’intégrité de la membrane mitochondriale correspond au point de non-retour lors de l’induction de l’apoptose, car elle conduit à une amplification du signal de mort vers les protéines effectrices. D’autre part, dans la plupart des cas, la voie extrinsèque d’induction de l’apoptose conduit à l’activation de la voie intrinsèque (Yin et al., 1999). La voie intrinsèque est considérée comme la voie maîtresse de l’induction de l’apoptose, de par son positionnement à la convergence de multiples voies de régulation de la survie et de la mort cellulaires.
Figure 1.1 – Voies de signalisation induisant l’apoptose classique. La voie extrinsèque est
activée par la liaison d’un ligand à un récepteur de mort membranaire. La voie intrinsèque est stimulée par un stress intracellulaire via l’activation de p53. Ces deux voies convergent vers l’activation des caspases, c’est-à-dire le clivage des procaspases en caspases actives. Les caspases clivent leurs protéines cibles, notamment des caspases effectrices. L’amplification du signal apoptotique mène à la perméabilisation de la membrane de la mitochondrie et la
5
relâche cytoplasmique de facteurs menant à la fragmentation du génome et au bourgeonnement membranaire.
La dérégulation des mécanismes de mort cellulaire programmée est liée à plusieurs classes de pathologies, dont les maladies neurodégénératives, l’immunodéficience, les maladies auto-immunes, ainsi que le cancer. En effet, l’acquisition de mutations rendant les cellules insensibles à l’induction de la mort cellulaire programmée fait partie des caractéristiques principales (hallmarks) du cancer telles qu’énoncées par Hanahan et Weinberg (Hanahan et Weinberg, 2000 ; Hanahan et Weinberg, 2011). Dans un contexte physiologique, l’une des fonctions de l’apoptose est d’éliminer les cellules anormales qui pourraient avoir un effet délétère pour l’homéostasie de l’organisme. L’évasion à l’apoptose permet aux cellules transformées de proliférer même en présence de signaux inhibiteurs de croissance ou de signaux de mort cellulaire. L’identification de voies alternatives de mort cellulaire programmée présente donc un intérêt majeur pour la recherche contre le cancer. L’étude de ces mécanismes pourrait permettre de développer des thérapies exploitant des voies non canoniques de mort cellulaire auxquelles les cellules cancéreuses demeureraient sensibles.
1.1.3 Mécanismes de mort cellulaire non apoptotiques
Le terme de « mort cellulaire non apoptotique » désigne un processus de mort cellulaire ne nécessitant pas l’activation des caspases et/ou ne présentant pas les caractéristiques morphologiques classiques de l’apoptose, mais qui peut être contrôlé au niveau cellulaire (Leist and Jäättelä, 2001).
À la fin des années 1990, l’avènement des méthodes de génétique murine a permis d’élaborer les premiers modèles dépourvus de gènes impliqués dans l’apoptose. De façon surprenante, la délétion de gènes pro-apoptotiques tels que Bax et Bak provoque l’apparition de syndactylie et une mortalité périnatale importante ; cependant, les individus qui survivent peuvent vivre plusieurs mois, indiquant que l’homéostasie de l’organisme est maintenue (Knudson et al., 1995 ; Lindsten et Thompson, 2006).
L’hypothèse proposée pour expliquer ce paradoxe fut alors l’existence de processus de mort cellulaire programmée non apoptotiques, c’est-à-dire ne dépendant pas des caspases. Cette
6
hypothèse fut ensuite validée par l’étude de l’apoptose des cellules interdigitales lors du développement de la souris. Ce processus représente l’un des mécanismes d’apoptose classique, dépendant des caspases, les plus souvent cités. Cependant, il a été mis en évidence que l’inhibition pharmacologique des caspases ou la délétion du gène Apaf1 (codant une protéine adaptatrice activant les caspases via la voie mitochondriale) n’empêchent pas la mort des cellules interdigitales (Chautan et al., 1999). De façon intéressante, dans ce modèle d’inhibition de l’apoptose classique, les cellules semblent mourir par un processus qui ressemble morphologiquement à la nécrose (noyau marbré, chromatine partiellement décompactée). Cette étude représente la première preuve de l’existence de mécanismes de mort cellulaire non apoptotiques in vivo chez les mammifères, ainsi que la possibilité d’un phénomène de nécrose programmée. Depuis ces études initiales, la mort cellulaire non apoptotique a été observée dans de nombreux contextes (Cheng et al., 2001 ; (Candé et al., 2002);Krishna et Overholtzer, 2016; Weinlich et al., 2017 ; Kutscher et Shaham, 2017).
Les processus de mort cellulaire non apoptotiques sont régulés par des voies de signalisation intracellulaires, et sont définis par des caractéristiques biochimiques plutôt que morphologiques (Galluzzi et al., 2012). Cependant, la diversité des modes de mort cellulaire rend difficile leur classification en catégories parfaitement distinctes. En effet, les voies de signalisation communiquent entre elles, de telle façon que l’activation d’un effecteur biochimique n’est pas suffisante pour conclure sur la nature du processus de mort (Kroemer et al., 2009). D’autre part, il a été établi qu’un même stimulus peut induire différents modes de mort selon la lignée cellulaire ou le contexte physiologique (Hirsch et al., 1997 ; Scheller et al., 2006). En outre, plusieurs voies de mort cellulaire peuvent être activées de façon concomitante et provoquer un phénotype mixte (Golstein et Kroemer, 2005). Ce phénomène repose notamment sur la perméabilisation de la membrane mitochondriale en tant que point de convergence des voies d’induction de la mort cellulaire. Étant donné leur grande diversité, seuls quelques mécanismes de mort non apoptotiques pertinents dans le cadre de cette thèse seront détaillés ici.
7
1.1.3.1 L’apoptose non classique
Les mécanismes de mort cellulaire regroupés sous le nom d’apoptose non classique sont morphologiquement semblables à l’apoptose mais ne dépendent pas de l’activation des caspases. Les cellules qui meurent par ces processus peuvent présenter certaines caractéristiques de cellules apoptotiques, telles que la condensation du corps cellulaire et du noyau par exemple. La mort induite par le facteur E4orf4 de l’adénovirus humain, qui fait l’objet de cette thèse, entre dans la catégorie des processus d’apoptose non classique dans certains modèles cellulaires.
1.1.3.2 La mort cellulaire par autophagie
L’autophagie (dénomination usuelle du mécanisme de macroautophagie) est un processus conservé de dégradation de composantes cellulaires, par la formation de vésicules à double membrane qui engouffrent des protéines ou des organelles entières. Ces vésicules appelées autophagosomes fusionnent ensuite avec le lysosome pour conduire à la dégradation protéolytique puis au recyclage de leur contenu (voir la section 1.5.1.2 pour plus de détails). La mort cellulaire par autophagie a été identifiée en 1973 comme l’un de trois types de mort cellulaire, avec l’apoptose et la nécrose (Schweichel et Merker, 1973).
La mort cellulaire par autophagie est présumée résulter d’un excès de flux autophagique. Morphologiquement, elle se manifeste par une dégradation des organelles mais une préservation des éléments du cytosquelette et une absence de réponse inflammatoire. Les cellules mourant par autophagie présentent de nombreuses vacuoles. Il a été montré que l’inhibition pharmacologique ou génétique de l’autophagie réduisait la mort cellulaire. La 3-méthyladénine, un nucléotide inhibiteur de la kinase PI3K et de l’autophagie, inhibe partiellement la mort des cellules de carcinome mammaire humaines traitées aux anti-œstrogènes (Bursch et al., 1996), ainsi que dans plusieurs autres contextes (Xue et al., 1999 ; Canu et al., 2005 ; Jia et al., 1997). Cependant dans ces études, il est difficile de déterminer dans quelle mesure l’autophagie est un processus de mort, ou en réalité un stimulus induisant l’apoptose. En effet, de nombreuses voies de stress cellulaire activent à la fois l’autophagie et l’apoptose. Par exemple, les protéines anti-apoptotiques de la famille Bcl-2 interagissent avec Beclin-1, protéine impliquée dans la nucléation de l’autophagosome, et cette interaction
8
inhibe l’induction de l’autophagie (Pattingre et al., 2005). Les protéines de la famille de Bcl-2 contrôlent la voie apoptotique mitochondriale et l’autophagie, et constituent un nœud de signalisation entre ces deux voies pouvant mener à une mort cellulaire impliquant l’autophagie (Gump et Thorburn, 2011). Il existe de nombreuses autres connexions entre les voies de régulation de l’autophagie et de la mort cellulaire, qui permettent la formation de réseaux maintenant l’homéostasie cellulaire. L’ensemble des résultats expérimentaux suggère un modèle dans lequel un stress cellulaire modéré stimulerait l’autophagie en tant que mécanisme protecteur. Toutefois, la persistance du stress conduirait finalement à l’accumulation de dommages cellulaires et à l’induction de la mort cellulaire (Mariño et al., 2014).
Des études plus récentes ont permis d’établir la première preuve génétique que l’autophagie pouvait induire la mort cellulaire. En effet, la déplétion par siARN des inducteurs autophagiques ATG7 et Beclin-1 bloque la mort des cellules murines L929 traitées avec l’inhibiteur de caspases zVAD-FMK (Yu et al., 2004). De plus, la déplétion de la protéine ATG5 (impliquée dans l’autophagie) et de Beclin-1 inhibe la mort des fibroblastes embryonnaires de souris Bax-/- ; Bak-/- traités à la staurosporine ou à l’étoposide (Shimizu et
al., 2004). Ces données prouvent que l’autophagie peut conduire à la mort sans déclencher l’apoptose, mais ne démontrent pas que cette voie de mort se déroule quand la machinerie apoptotique est intacte.
Néanmoins, l’autophagie est généralement considérée comme un mécanisme facilitant la survie cellulaire en cas de stress, puisqu’elle assure une source alternative de précurseurs de composants cellulaires. De plus, elle protège la cellule en dégradant les organelles défectueuses qui pourraient avoir un effet néfaste sur la survie : elle assure ainsi un contrôle de qualité (Anding et Baehrecke, 2017). Il existe notamment un mécanisme d’autophagie sélective visant à dégrader les mitochondries dysfonctionnelles, par le processus de mitophagie. Les mitochondries endommagées représentent un danger pour les cellules, par leur impact sur les espèces réactives de l’oxygène ou leur susceptibilité à se perméabiliser de façon incontrôlée (Lemasters, 2005). D’autres processus d’autophagie sélective assurent l’intégrité du réticulum endoplasmique, de la membrane plasmique et de protéines
9
endommagées par les stress cellulaires, afin de limiter leur effet délétère sur la survie cellulaire (Green et Levine, 2014). Toutefois, les mécanismes moléculaires assurant la spécificité de ces voies de dégradation sont encore peu compris ; le rôle émergeant des chaperons moléculaires sera discuté à la section 1.5.
1.1.4 Les protéines tumoricides
L’hétérogénéité des modes de mort cellulaire programmée illustre la diversité des voies de signalisation induisant la mort, ainsi que l’importance de leur régulation pour le fonctionnement normal de l’organisme. Ces sentiers signalétiques forment des réseaux complexes finement régulés. En outre, les protéines régulant l’induction normale de l’apoptose sont fréquemment dérégulées ou mutées dans les cellules cancéreuses, ce qui limite l’efficacité des traitements anti-cancéreux ciblant ces voies classiques (Lowe et al., 2004 ; Adams et Cory, 2007 ; Sharma et Settleman, 2010). L’identification de voies alternatives de mort cellulaire auxquelles les cellules transformées seraient communément sensibles permettrait alors de développer des traitements efficaces contre une grande variété de tumeurs.
Dans ce contexte, les protéines tumoricides représentent des modèles de choix. Une protéine est dite tumoricide si son expression induit la mort cellulaire de façon préférentielle dans les cellules transformées, par rapport aux cellules normales (Bruno et al., 2009). A ce jour, plusieurs protéines tumoricides ont été décrites, parmi lesquelles on retrouve des protéines virales (apoptine, E1A, NS1), cellulaires (TRAIL, MDA7, Brevinin-2R) et du lait maternel humain (HAMLET) (Backendorf et Noteborn, 2014). La protéine E4orf4 de l’adénovirus humain est considérée comme l’une d’entre elles.
L’identification des protéines cellulaires impliquées dans l’effet tumoricide de ces protéines est un enjeu majeur de recherche. En effet, elles feraient partie de voies de signalisation modifiées par la transformation oncogénique, partagées par la plupart des cellules transformées, et qui les rendraient sensibles à leur activité toxique. D’autre part, puisque ces voies alternatives semblent prédominer pour induire la mort des cellules transformées, elles pourraient interagir fonctionnellement avec la signalisation oncogénique. De cette façon,
10
l’acquisition du caractère transformé par une cellule se ferait conjointement avec l’émergence de voies non apoptotiques comme uniques modes de mort cellulaire. Ainsi, l’identification des effecteurs impliqués dans les processus tumoricides pourrait permettre de mettre en lumière les régulateurs centraux de la signalisation oncogénique.
Tableau 1.1 – Caractéristiques des principales protéines tumoricides connues. Protéine Origine Localisation
subcellulaire Mécanisme de mort cellulaire Dépendance envers p53 Bcl-2 Statut thérapeutique Apoptine Virus de l’anémie du poulet Cytoplasme (cellules normales), noyau (cellules cancéreuses) Voie apoptotique intrinsèque
Non Non1 Études
précliniques HAMLET Complexe de l’α-lactalbumine du lait humain et l’acide oléique Cytoplasme (cellules normales), noyau (cellules cancéreuses) Apoptose,
autophagie Non Non Études cliniques de phase I et II TRAIL Ligand de récepteurs de la famille du TNF Liaison à un récepteur extracellulaire Voie apoptotique extrinsèque Non ? Études cliniques de phase I et II MDA7 Cytokine de la famille IL-10 Liaison à un récepteur extracellulaire Voie apoptotique extrinsèque
Non Oui2 Études
cliniques de phase I et II E4orf4 Adénovirus
de type 2 Noyau (précoce), cytoplasme (tardif)
Apoptose non classique
Non Non Études précliniques
NS1 Virus
minute de la souris (parvovirus)
Cytoplasme Autophagie Non3 ? Études
précliniques
Brevinine-2R Grenouille Rana ridibunda
Cytoplasme
(lysosomes) Autophagie ? Oui
4 Études
précliniques
Bruno et al., 2009 ; 1 Danen-Van Oorschot et al., 1999 ; 2 Lebedeva et al., 2003 ; 3 Mincberg
11
Des études précliniques sont en cours pour développer des approches thérapeutiques utilisant l’apoptine, par thérapie génique ou protéique, afin de cibler les cellules cancéreuses. De plus, des thérapies combinées ont mis en évidence que l’apoptine stimule la sensibilité des cellules cancéreuses à l’action de certains traitements de chimiothérapie dans un modèle murin, permettant d’en réduire les doses et les effets secondaires (Jin et al., 2011). La protéine HAMLET a été testée lors d’études précliniques sur des patients humains et a démontré une efficacité thérapeutique dans des cas de cancers de la peau et de la vessie (Gustafsson et al., 2004; Mossberg et al., 2007). Tous ces éléments soulignent l’intérêt de l’étude des protéines tumoricides pour, d’une part, acquérir une meilleure compréhension des voies de survie cellulaire dans les cellules transformées, et améliorer l’efficacité des traitements de chimiothérapie d’un point de vue clinique.
De façon générale, la mort cellulaire induite par les protéines tumoricides n’est pas affectée par l’utilisation d’inhibiteurs de caspases, et elle est indépendante de Bcl-2 et de p53. La signalisation de Bcl-2 et de p53 est dérégulée dans la majorité des cancers, ce qui contribue à l’évasion des cellules transformées aux signaux apoptotiques classiques (Adams et Cory, 2007 ; Duffy et al., 2014). En induisant la mort cellulaire indépendamment de ces voies canoniques, les protéines tumoricides assureraient leur effet toxique dans les cellules transformées.
1.2 La protéine E4orf4 de l’adénovirus 1.2.1 E4orf4 et la réplication virale
Les adénovirus humains sont responsables d’infections des voies respiratoires (Wold et Horvitz, 2007). Ce sont des virus à ADN non enveloppés qui infectent les cellules épithéliales. Leur génome est constitué d’un ADN double brin linéaire d’environ 36 kb et présente des gènes précoces (E) et tardifs (L) (Cheng et al., 2015).
Lors de l’infection, l’adénovirus se fixe aux récepteurs CAR (Coxsackie and Adenovirus
Receptor) à la surface de la cellule, avant de pénétrer dans le cytoplasme par endocytose
dépendante de la clathrine (Bergelson et al., 1997 ; Wickham et al., 1993). Le virus est transporté via les microtubules jusqu’au noyau, où les gènes précoces sont transcrits (Strunze
12
et al., 2011). La protéine virale précoce E1A est un facteur de transcription qui stimule l’expression des gènes de la cellule-hôte impliqués dans la réplication de l’ADN, afin de faciliter la copie du génome viral (Whyte et al., 1988 ; Stabel et al., 1985 ; Kaczmarek et al., 1986). Ce passage aberrant en phase S active p53, qui est inhibée par la protéine virale E1B 55K (Yew et Berk, 1992). De la même façon, la protéine E1B 19K, homologue de Bcl-2, consolide l’inhibition de l’apoptose pour permettre la poursuite de la réplication virale (Rao et al., 1992). Ainsi, l’infection virale représente un contexte similaire à la transformation oncogénique, puisqu’E1A favorise la progression dans le cycle cellulaire et que les protéines E1B inhibent l’induction de l’apoptose. E4orf4 exerce donc ses fonctions virales dans des conditions signalétiques semblables à la transformation tumorale.
La région E2 du génome viral code 3 protéines qui régulent la réplication du génome viral (Caravokyri et Leppard, 1996 ; de Jong et al., 2003). Les gènes de la région E3 modulent la réponse antivirale cellulaire de l’hôte et induisent la mort cellulaire à l’issue de l’infection (Tollefson et al., 1996 ; Zou et al., 2004;Fessler et al., 2004). Les produits de la région E4 présentent des fonctions diverses telles que la régulation de l’expression des gènes viraux et le transport des ARNm hors du noyau (Weinberg et Ketner, 1986 ; Dix et Leppard, 1993). Certains gènes de la région E4 ont également un rôle dans l’induction de la mort des cellules infectées par un processus indépendant de p53 (Täuber and Dobner, 2001). En particulier, l’étude individuelle des protéines codées par les gènes de la région E4 a permis de démontrer que la protéine E4orf4 présente une activité toxique (Lavoie et al., 1998 ; Shtrichman et Kleinberger, 1998 ; Marcellus et al., 1998). Enfin, les gènes de la région L codent des protéines structurales qui s’assemblent autour du génome viral pour former des virions, complétant ainsi le cycle de réplication du virus ((Mangel and San Martín, 2014) ; Cheng et al., 2015).
Il a été montré que lors de la réplication, E4orf4 induit une baisse d’expression des protéines virales précoces et des gènes cellulaires influençant la réplication virale (Ben-Israel et al., 2008; Bondesson et al., 1996 ; Mannervik et al., 1999 ; (Müller et al., 1992)). Elle régule également la réplication du génome viral (Bridge et al., 1993 ; Medghalchi et al., 1997), l’épissage des ARNm viraux (Estmer Nilsson et al., 2001 ; Kanopka et al., 1998) et la
13
traduction des protéines ((Müller et al., 1992) ; (O’Shea et al., 2005)). E4orf4 n’est pas indispensable à la réplication virale ; sa délétion dans le génome viral conduit à un ralentissement du cycle de réplication (Halbert et al., 1985). Les fonctions précises d’E4orf4 ainsi que de ses partenaires cellulaires dans le contexte de l’infection demeurent peu connus.
1.2.2 Action tumoricide d’E4orf4
L’étude fonctionnelle systématique de la région E du génome viral a mis en évidence que la région E4 était impliquée dans l’induction de l’apoptose par la protéine E1A, de façon indépendante de p53 (Marcellus et al., 1996). En effet, les cellules infectées avec des adénovirus mutants délétés de la région E4 demeurent viables plus longtemps que les cellules infectées par les virus contrôle. Les études subséquentes ont révélé que l’expression de la protéine E4orf4, mais pas celle des autres protéines de la région E4, était suffisante pour induire la mort cellulaire, identifiant alors E4orf4 comme le facteur toxique de la région E4 (Marcellus et al., 1998). Ainsi, dans le contexte de la réplication virale, les fonctions d’E4orf4 participent à l’induction de la mort cellulaire, et cette activité se manifeste également lorsque la protéine est exprimée seule.
Il a été établi que la mort induite par E4orf4 présente des caractéristiques morphologiques de l’apoptose (condensation nucléaire, dysfonctions mitochondriales) dans plusieurs lignées cellulaires transformées et qu’il s’agit d’un processus indépendant de l’activation des caspases (Lavoie et al., 1998). Il a toutefois été observé dans plusieurs lignées cellulaires transformées ainsi que chez la drosophile que les caspases pouvaient être activées en aval d’E4orf4 (Livne et al., 2001 ; Robert et al., 2002 ; Pechkovsky et al., 2013). Ce phénomène est considéré comme un mécanisme d’amplification de la signalisation de mort cellulaire. De plus, l’inhibition pharmacologique des caspases n’inhibe pas la mort cellulaire induite par E4orf4, mais conduit dans certains cas à l’apparition de phénotypes de mort différents (Robert et al., 2002). Cela souligne la régulation croisée (crosstalk) des voies de signalisation conduisant à la mort cellulaire, ici dans le cas particulier d’E4orf4.
De façon intéressante, la transformation oncogénique sensibilise les cellules primaires à l’activité toxique d’E4orf4 (Shtrichman et Kleinberger, 1998 ; Shtrichman et al., 1999). En
14
effet, l’expression des oncogènes Myc ou Ras dans les cellules primaires de rein de bébé rat est suffisante pour doubler le nombre de cellules apoptotiques en présence d’E4orf4. Des résultats non publiés de notre laboratoire indiquent que l’induction de la kinase oncogénique Src dans les cellules MDCK (Madin-Darby canine kidney cells, cellules épithéliales de rein de chien, non transformées) est suffisante pour les sensibiliser à l’activité toxique d’E4orf4. Ces expériences soulignent le rôle central de Src dans l’activité tumoricide d’E4orf4 (discuté à la section 1.2.3.1). En outre, il a été mis en évidence par notre équipe et d’autres que l’expression d’E4orf4 dans de nombreuses lignées de cellules transformées ou cancéreuses mène à la mort cellulaire, tandis qu’elle a peu d’effet sur la viabilité des cellules non transformées (Shtrichman et al., 1999 ; chapitre 2 de cette thèse). Tous ces éléments contribuent au modèle selon lequel E4orf4 présente une activité tumoricide, c’est-à-dire que son activité toxique se manifeste dans les cellules transformées mais épargne les cellules normales. La protéine E4orf4 induit un mode d’apoptose indépendant des caspases et de p53, résistant à la surexpression de Bcl-2, ce qui en fait un outil thérapeutique potentiellement puissant pour contrer le cancer. Il est donc essentiel d’identifier les protéines cellulaires interagissant fonctionnellement avec E4orf4 et les mécanismes moléculaires impliqués dans son activité tumoricide.
Il est important de noter qu’une différence essentielle existe entre le contexte de réplication virale, c’est-à-dire le contexte physiologique de l’expression d’E4orf4, et son effet toxique dans les cellules transformées. En effet, les analyses de l’effet d’E4orf4 hors du contexte viral reposent sur sa surexpression après transfection, menant à un niveau d’expression d’E4orf4 bien supérieur à celui retrouvé dans les cellules infectées par l’adénovirus (Mui et al., 2013). D’ailleurs lors de l’infection, E4orf4 régule à la baisse sa propre expression via une boucle de rétrocontrôle transcriptionnel (Bondesson et al., 1996). E4orf4 est une protéine d’échafaudage qui interagit avec les protéines cellulaires via ses domaines d’interaction. Lorsqu’elle est surexprimée dans les cellules transformées, son abondance pourrait conduire à la titration de certaines de ses protéines cibles en les empêchant d’accomplir leurs fonctions. À l’inverse lors de l’infection virale, son niveau d’expression modéré pourrait rediriger une plus petite fraction du pool de ses protéines cibles, en interférant peu avec leurs fonctions physiologiques. Selon ce modèle, certains mécanismes d’action d’E4orf4 lors de l’infection
15
virale et dans l’induction de la mort des cellules transformées seraient partagés, mais pas totalement identiques.
1.2.3 Signalisation de la mort cellulaire induite par E4orf4
E4orf4 est une protéine de 14 kDa qui ne possède pas de domaine catalytique connu. Les outils informatiques de prédiction de structure des protéines suggèrent qu’E4orf4 pourrait être constituée de 3 hélices alpha et de boucles en ses extrémités N- et C-terminales (Horowitz et al., 2013). Le modèle actuel propose qu’E4orf4, comme beaucoup de protéines virales, accomplirait ses fonctions en s’associant avec des protéines cellulaires afin de subvertir leur activité.
Les deux cibles cellulaires d’E4orf4 les mieux caractérisées sont les kinases de la famille de Src (Src family kinases ou SFK) (Lavoie et al., 2000; Champagne et al., 2004) et la sous-unité Bα de la phosphatase de sérines et thréonines PP2A (Kleinberger et Shenk, 1993 ; Shtrichman et al., 2000 ; Marcellus et al., 2000). Les kinases Src présentent un rôle oncogénique alors que la phosphatase PP2A est considérée comme un suppresseur de tumeur ; leur interaction avec E4orf4 est donc probablement très pertinente pour l’activité tumoricide de la protéine virale. Les domaines d’E4orf4 qui permettent son interaction avec Src et PP2A ont été identifiés et se chevauchent partiellement (Champagne et al., 2004 ; Marcellus et al., 2000). Toutefois, la mutation F84A, qui se situe dans le domaine de liaison à PP2A, induit une perte d’interaction avec PP2A tout en conservant la liaison à Src (Marcellus et al., 2000 ;Champagne et al., 2004). Ce mutant démontre un gain de fonction dans l’activité toxique dépendant de Src (Champagne et al., 2004). Cet élément, entre autres, indique que Src et PP2A sont probablement impliqués dans la mort cellulaire par des processus distincts, dont la contribution relative serait variable selon le contexte cellulaire.
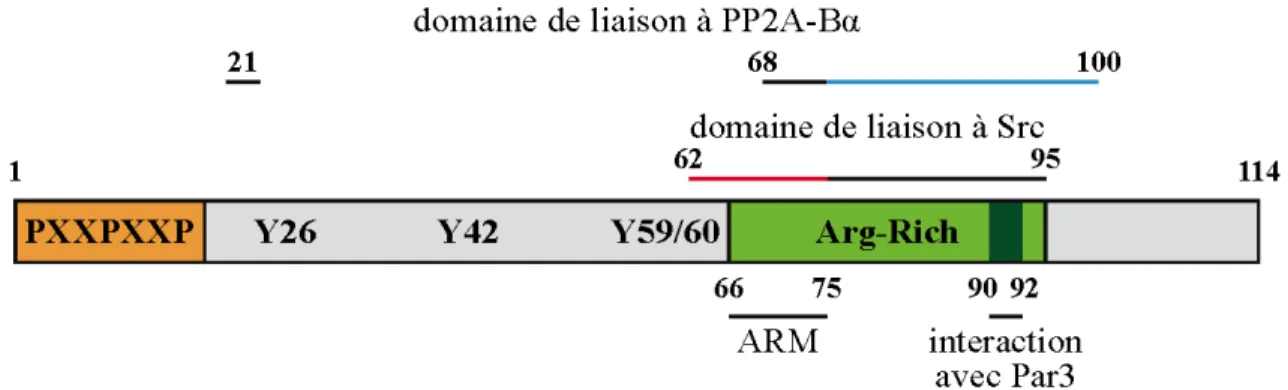
E4orf4 présente un domaine riche en prolines (PXXP) à son extrémité N-terminale qui permet son association avec des protéines à domaine SH3 (Src homology 3) in vitro (Figure 1.2) (Gingras et al., 2002 ; données non publiées). Cependant, les protéines cellulaires qui lieraient ce domaine et contribueraient à la mort cellulaire n’ont pas été identifiées. Quatre résidus tyrosine ont été identifiés comme étant phosphorylés par les kinases Src lors de
16
l’induction de la mort cellulaire (en positions 26, 42, 59 et 60). En effet, leur mutation en résidus non-phosphorylables (mutant 4YF) inhibe l’induction de la mort cellulaire dépendante de Src (Gingras et al., 2002). Enfin, E4orf4 possède un motif riche en arginines dans sa moitié C-terminale qui sert de signal de localisation nucléaire et nucléolaire (Miron et al., 2004). Ce domaine est impliqué dans la localisation subcellulaire d’E4orf4 et participe à la régulation de son activité toxique. En effet, sa dynamique entre le noyau et le cytoplasme régule l’induction de la mort dans les cellules transformées (chapitre 2 ; Gingras et al., 2002).
Figure 1.2 – Schéma de la structure de la protéine E4orf4 de l’adénovirus. La taille relative
des domaines est respectée. ARM = arginine-rich motif, motif de localisation nucléaire et nucléolaire.
1.2.3.1 Rôle de Src
Les kinases oncogéniques de la famille Src
Les kinases de la famille Src sont des tyrosine kinases qui ne sont pas des récepteurs. Src est le prototype de cette famille, dont les deux membres les plus proches sont Fyn et Yes. Ces trois protéines sont presque ubiquitaires dans l’organisme ; d’autres sont exprimées surtout dans les cellules hématopoïétiques (Lck, Hck, Blk, Fgr et Lyn). Les kinases Src sont activées par de nombreux stimuli tels que les récepteurs tyrosine kinase, les récepteurs couplés aux protéines G et les intégrines. Leurs cibles sont impliquées dans la prolifération, l’apoptose, le contrôle des cytosquelettes et dans l’adhésion cellulaire (Thomas et Brugge, 1997 ; Yeatman, 2004 ; Summy et Gallick, 2006).
Les kinases Src sont des oncogènes et sont fréquemment retrouvées suractivées dans les cellules cancéreuses, corrélant avec un mauvais pronostic (Aligayer et al., 2002 ; Matsumoto