DOCTEUR DE L'UNIVERSITÉ DE POITIERS UFR de médecine et de pharmacie
Laboratoire de neurosciences expérimentales et cliniques - LNEC (Poitiers) (Diplôme National - Arrêté du 25 mai 2016)
École doctorale : Biologie-santé - Bio-santé (Limoges)
Secteur de recherche : Aspects Moléculaires et Cellulaires de la Biologie
Présentée par :
Konstantin Masliantsev
Rôle des signalisations STAT3 et Hippo dans les gliomes : Identification de nouveaux biomarqueurs pronostiques
et cibles thérapeutiques
Directeur(s) de Thèse : Lucie Karayan-Tapon
Soutenue le 04 décembre 2018 devant le jury Jury :
Président Pierre Verrelle Professeur et praticien hospitalier, Clermont-Ferrand
Rapporteur Ahmed Idbaih Professeur et praticien hospitalier, CHU Pitié Salpétrière
Rapporteur Corinne Auge-Gouillou Professeur, Université de Tours
Membre Lucie Karayan-Tapon Professeur et praticien hospitalier, Université de Poitiers
Membre Michel Wager Professeur et praticien hospitalier, Université de Poitiers
Membre Omar Benzakour Professeur, IPBC, Université de Poitiers
Membre Philippe Arnaud Professeur, GReD, Université de Clermont-Ferrand
Membre Fabrice Lalloué Professeur, Université de Limoges
Pour citer cette thèse :
Konstantin Masliantsev. Rôle des signalisations STAT3 et Hippo dans les gliomes : Identification de nouveaux
biomarqueurs pronostiques et cibles thérapeutiques [En ligne]. Thèse Aspects Moléculaires et Cellulaires de la
DOCTEUR DE L’UNIVERSITE DE POITIERS (Faculté Médecine et Pharmacie) (Diplôme National - Arrêté du 25 mai 2016)
Ecole Doctorale : Biosanté n° 524
Secteur de Recherche : Aspects Moléculaires et Cellulaires de la Biologie Présentée par :
Konstantin MASLIANTSEV
************************
Rôle des signalisations STAT3 et Hippo dans les gliomes :
Identification de nouveaux biomarqueurs pronostiques
et cibles thérapeutiques
************************Directeur de Thèse : Pr Lucie KARAYAN-TAPON Soutenue le 4 Décembre 2018 devant la Commission d’Examen
************************
JURY
Rapporteurs : Pr Corinne AUGE-GOUILLOU Dr Ahmed IDBAIH
Examinateurs : Dr Philippe ARNAUD Pr Omar BENZAKOUR Pr Lucie KARAYAN-TAPON Pr Fabrice LALLOUE
Pr Pierre VERRELLE Pr Michel WAGER
Tout d’abord je voudrais remercier le Pr Corinne Augé-Gouillou et le Dr Ahmed Idbaih d’avoir accepté d’être les rapporteurs de mon travail de thèse. Je tiens également à remercier le Dr Philippe Arnaud, Pr Omar Benzakour, Pr Fabrice Lalloué, Pr Pierre Verrelle et le Pr Michel Wager pour avoir accepté de faire partie de mon jury de thèse.
Je tiens à remercier particulièrement le Pr Lucie Karayan-Tapon de m’avoir accueilli au sein de son groupe depuis mon stage de Master 2 et de m’avoir permis d’effectué cette thèse. Je vous remercie sincèrement pour la confiance que vous m’avez accordée.
Je remercie le Pr Mohamed Jaber, directeur du Laboratoire de Neurosciences Expérimentales et Cliniques – INSERM U1084, ainsi que le Pr Afsaneh Gaillard responsable de l’Equipe 1 : Thérapies cellulaires dans les pathologies cérébrales de m’avoir accueilli au sein du LNEC durant ma thèse.
Je remercie chaleureusement l’association « En Avant la Vie » pour votre soutien financier et le travail que vous faites au quotidien auprès de malades.
Je tiens particulièrement à remercier le Dr Pierre-Olivier Guichet pour ces trois ans passé à travailler ensemble. Je te remercie pour ton aide précieuse tout au long de cette thèse. Pour moi tu es à la fois un mentor et un ami, c’est toi qui m’as tout appris.
Je tiens à remercier le Dr Véronique Ladevèze (vous êtes un peu ma maman dans la recherche) et le Dr Anaïs Balbous (tu es un peu ma grande sœur dans la recherche) pour m’avoir toujours encouragé et m’avoir incité à poursuivre en thèse.
Je voudrais également remercier tous les professeurs que j’ai eus durant mon cursus universitaire et plus particulièrement durant les années du Master pour avoir essayé de nous apprendre à réfléchir et donner l’envie de faire de la recherche.
Je voudrais aussi remercier les membres du « neuro-club » pour m’avoir permis d’avoir une plus large ouverture d’esprit et m’avoir apporté un regard plus clinique sur la maladie.
plaisir), le LITEC pour la caméra de révélation de mes Westerns où j’ai passé un petit moment, et Adriana pour les manips de FACS.
Je remercie sincèrement tous les membres du LNEC et du Labo de Cancéro-Bio pour votre aide, vos encouragements et les bons moments passé ensemble. Merci à Gaëlle, Pierre, Seb, Tristan, Gwen, Claire, Ulrich, Marie-Laure, Valérie et tous ceux que j’ai oublié. Merci également aux anciens membres, Christos et Amir, pour votre soutien et les cafés au distributeur.
Merci à tous mes collègues et amis doctorants et post-docs : JoJo, Aurélie, Thomas, Hugo, Obé, Adélie, Manu, MH et tous ceux que j’oublie pour tous les bons moments passés ensemble à refaire le monde et la science ou les moins bons quand il fallait s’encourager mutuellement pour continuer et aller plus loin.
Merci à mes amis qui m’ont toujours soutenu : LioLio et TiTi depuis toujours, Thomas depuis les années Fac et tous les autres.
Au tour de la Famille maintenant…Je remercie mes parents qui m’ont donné le goût pour les sciences. Je n’oublierai jamais la première fois quand mon Père m’a fait regarder des cellules au microscope (je devais avoir 5 ans) et que j’ai découvert avec stupeur qu’elles étaient rondes, alors que je m’attendais à voir des beaux carreaux comme dans un cahier…Je remercie particulièrement mon beau-père Philippe et ma Mère sans qui jamais je n’aurais pu faire ces études. Merci à mes grands-parents pour m’avoir toujours encouragé sans jamais douter de moi. Je remercie toutes les personnes de ma Famille, que ce soit ma Famille Française ou Russe, vous m’avez tous soutenu durant ces années d’études. Je tiens à remercier également ma Belle Famille : Lolo et Marco, les Grands et les Petits, merci pour votre soutien. Merci à ma Clémence pour m’avoir supporté tous les jours depuis le commencement des études jusqu’au bout de la thèse, sans toi je ne serais pas arrivé là.
Je voudrais remercier toutes les personnes qui m’ont encouragé depuis l’Espagne à la Nouvelle Calédonie, survolant la Sibérie, en passant par Toulouse et Moscou, en faisant une escale à Angoulême et Samara et atterrissant finalement ici à Poitiers.
LISTE DES ABRÉVIATIONS ... 1
TABLE DES ILLUSTRATIONS ... 4
INTRODUCTION ... 7
Chapitre I :Les Gliomes ... 9
Les tumeurs cérébrales ... 11
I. 1. Epidémiologie ... 11
2. Facteurs de risques ... 11
3. Symptômes et diagnostic ... 13
Classifications histologiques des gliomes ... 14
II. 1. Historique ... 14
2. Classification de l’OMS 2007 ... 16
1) Les astrocytomes ... 17
2) Les oligodendrogliomes ... 20
3) Les tumeurs mixtes ... 22
3. Classification de l’hôpital St Anne ... 23
Classification de l’OMS 2016 ... 24
III. 1. Statut IDH ... 25
2. Codélétion 1p19q ... 27
3. Autres marqueurs moléculaires ... 28
Les glioblastomes ... 29
IV. 1. Caractéristiques des GBM ... 29
2. Classification de Philipps ... 30
3. Classification de Verhaak ... 31
Les traitements des gliomes ... 33
V. 1. Chirurgie ... 33 2. Radiothérapie ... 34 3. Chimiothérapie ... 34 4. Autres traitements ... 35 1) Les nitrosourées ... 35
2) Le traitement ciblant les récepteurs tyrosine-kinases (RTK) ... 35
3) Le traitement anti-angiogénique ... 36
3. Origines ... 43
Hétérogénéité des CSG ... 45
II. Facteurs de régulation des CSG ... 46
III. 1. Les facteurs intrinsèques ... 47
1) Facteurs génétiques et épigénétiques ... 47
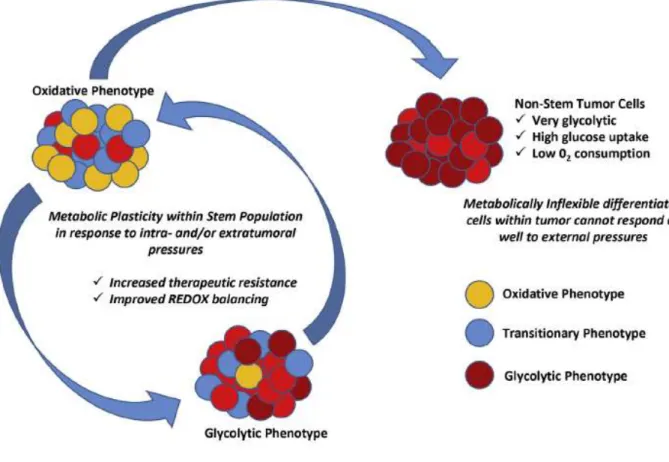
2) Métabolisme ... 49
2. Les facteurs extrinsèques ... 51
1) Niche et microenvironnement tumoral ... 51
2) Système immunitaire ... 54
Résistance aux traitements ... 54
IV. 1. Radiorésistance ... 55
2. Chimiorésistance ... 56
Les voies de signalisation impliquées dans le maintien des CSG ... 58
V. 1. NOTCH ... 58
2. WNT ... 60
3. Sonic Hedgehog ... 61
Chapitre III : Voies de signalisation impliquées dans la pathogenèse des gliomes ... 65
Les voies de signalisation en tant que cibles thérapeutiques ... 67
I. La voie de signalisation STAT3 ... 68
II. 1. Acteurs de la signalisation STAT3 ... 68
1) Facteurs de transcription de la famille STAT ... 68
2) Signalisation STAT3 ... 69
3) Régulateurs de la voie STAT3 ... 71
2. STAT3 dans les gliomes ... 73
1) Mécanismes d’activation constitutive de STAT3 dans les cellules tumorales ... 74
2) Rôle de STAT3 dans les processus tumorigéniques et la résistance aux traitements ... 75
3) Valeur pronostique de l’activation de STAT3 ... 78
La voie de signalisation Hippo ... 80
III. 1. Acteurs de la signalisation Hippo ... 81
2. Rôle biologique ... 83
3. Rôle de la signalisation Hippo dans les cellules tumorales ... 85
4. Implication de la voie Hippo dans les tumeurs solides ... 88
5. Signalisation Hippo dans les gliomes ... 91
RT-qPCR ... 108
Western Blot ... 109
Fractionnement cellulaire ... 111
Immunoprécipitation ... 111
Immunofluorescence ... 111
Construction des Tissue MicroArray (TMA) ... 112
Immunohistochimie ... 112 Test de clonogénicité ... 113 Irradiation cellulaire ... 113 Cytométrie en flux ... 113 Analyses bioinformatiques ... 114 Statistiques ... 115 RÉSULTATS ... 117
Publication 1 : « Impact of STAT3 phosphorylation in Glioblastoma Stem Cells radiosensitization and patient outcome » ... 119
Publication 2 : « Fatal correlation between YAP1 expression and glioma aggressiveness: clinical and molecular evidence » ... 141
TRAVAUX EN COURS ... 171
Identification de TEAD3 en tant que facteur de mauvais pronostic dans les gliomes ... 173
Expression de TEAD3 dans les tissus de patients ... 176
Etat d’activation de la signalisation Hippo dans les CSG ... 178
Activation de TEAD3 dans les CSG ... 181
Rôle biologique de TEAD3 dans les CSG ... 183
DISCUSSION ET PERSPECTIVES ... 187
1
2-HG : 2-hydroxyglutarate
α-KG : α-cétoglutarate
ABC : ATP Binding Cassette
ABCB1 : ATP Binding Cassette Subfamily B Member 1
ADAM : A Disintegrin And Metalloproteinase
AD-HIES : syndrome hyper-IgE autosomique dominant
ADN : Acide désoxyribonucléique
AKT : Protein kinase B
AREG : an EGF-like growth factor
ARNm : Acide ribonucléique messager
ASCL1 : Achaete-Scute Family bHLH Transcription Factor 1
ATG5 : Autophagy Related 5
ATP : Adénosine triphosphate
ATRX : ATP-dependent helicase ATRX, X-linked helicase II
BCAN : Brevican
BCL-XL : B-cell lymphoma-extra large
BCNU : carmustine
BCR-ABL : Breakpoint Cluster Region-Abelson
BHE : Barrière Hémato-Encéphalique
BMX : Bone Marrow X-linked
CCNU : Lomustine
CD133 : Prominine-1
CDKN2A : Cyclin Dependent Kinase Inhibitor 2A
C/EBPβ : CCAAT/Enhancer-Binding Protein beta
CHI3L1 : Chitinase-3-like protein 1
CHK : Checkpoint Kinase
CLC : Cardiotrophin-Like Cytokine
Codel : codélétion 1p19q
CSC : Cellules Souches Cancéreuses
CSG : Cellules Souches de Glioblastomes
CSN : Cellules Souches Neurales
CTGF : Connective Tissue Growth Factor
CTLA-4 : Cytotoxic T-Lymphocyte-Associated molecules-4
CTNF : Ciliary Neurotrophic Factor
CYR61 : Cysteine Rich Angiogenic Inducer 61
DLL : Delta-like
EGF : Epidermal Growth Factor
EGFR : Epidermal Growth Factor Receptor
ERBB3 : Erb-B2 Receptor Tyrosine Kinase 3
bFGF : basic Fibroblast Growth Factor
FGFR : Fibroblast Growth Factor Receptor
FOXM1 : Forkhead Box M1
FZD : Frizzled
GABRA1 : Gamma-Aminobutyric Acid Type A Receptor Alpha1 Subunit
2
GAS1 : Growth arrest-specific protein 1
GBG : gliome de bas grade
GBM : Glioblastome
GFAP : Glial Fibrillary Acidic Protein
GLI : Zinc finger protein GLI
GSK3β : Glycogen Synthase kinase 3 beta
HDAC : Histone désacétylase
HIF : Hypoxia Inducible Factor
Hpo : Hippo
ICB : Immune Checkpoint Blockers
ID1 : Inhibitor Of DNA Binding 1
IDH1/2: Isocitrate déshydrogénases 1/2
IDHmut : IDH1/2 muté
IDHwt : IDH1/2 sauvage
IL : interleukine
IRM : imagerie par résonance magnétique
ITK : Inhibiteurs des Récepteurs à activité Tyrosine Kinase
JAG : Jagged
JAK : Janus Kinase
L1CAM : L1 Cell Adhesion Molecule
LATS1/2 : Large Tumor Suppressor Kinase 1/2
LIF : Leukemia Inhibitory Factor
LRP : LDL receptor related protein
MAP kinases : Mitogen-activated protein kinases
Reproductive Tyrosine Kinase
c-MET : Tyrosine-Protein Kinase Met
MGMT : 6-O-méthylguanine méthyltransférase
MLL1 : Mixed Lineage Leukemia 1
MMP : Matrix Metalloproteinases
MST1/2 : Mammalian Sterile20-like 1/2
NICD : Notch Intracellular Domain
NEFL : Neurofilament Light
NF : Neurofibromine
NF-κB : Nuclear Factor kappa-light-chain-enhancer of activated B cells
NHEJ : réparation par jonction d'extrémités non-homologues
NK : Natural Killer
NKX2.2 : NK2 homeobox 2
eNOS : Nitric Oxide Synthase 3
bHLH : basic Helix-Loop-Helix
Non-codel : absence de la codélétion 1p19q
OCT4 : Octamer-binding transcription factor 4
OLIG2 : Oligodendrocyte transcription factor 2
OMS : Organisation Mondiale de la Santé
OSM : Oncostatine M
PCNA : Proliferating Cell Nuclear Antigen
3
PI3K : Phosphoinositide 3-kinase
PIAS : Protein Inhibitor of Activated STAT3
PKC : Protéines Kinases C
PLAGL2 : Pleiomorphic adenoma gene-like 2
POU3F2 : POU Class 3 Homeobox 2
PP2A : Phosphoprotein Phosphatase 2A
PPAR-γ : Peroxisome proliferator-Activated Receptor-γ
PTCH1 : Patched1
PTEN : Phosphatase and Tensin Homolog PTPN : protein-tyrosine phosphatase non-receptor PTPR : receptor-type tyrosine-protein phosphatase RB1 : Retinoblastoma-associated protein
ROS : dérivés réactifs de l'oxygène
SALL2 : Spalt Like Transcription Factor 2
SAV1 : Salvador
SCID : Severe Combined Immunodefeciency
SH2 : Src Homology 2
SHh : Sonic Hedegehog
SLC12A5 : Solute Carrier Family 12 Member 5
Signaling
SOX2 : Sex Determining Region Y-Box 2
STAT3 : Signal Transducer And Activator Of Transcription 3
SUFU : Suppressor of fused
SVZ : zone subventriculaire
SYT1 : Synaptotagmin 1
TAZ : Transcriptional coactivator with PDZ-binding motif
TCGA : The Cancer Genome Atlas
TEAD : TEA domain
TERT : Telomerase Reverse Transcriptase
TMZ : Témozolomide
TNF : Tumor Necrosis Factor
TNFRSF1A : Tumor Necrosis Factor Receptor Superfamily Member 1A
TOP2A : DNA topoisomerase 2-alpha
TP53 : Tumor Protein 53
TRADD : Tumor necrosis factor receptor type 1-associated DEATH domain protein
VEGF : Vascular Endothelial Growth Factor
Wts : Warts
YAP1 : Yes-Associated Protein 1
Yki : Yorkie
ZFX : Zinc Finger Protein X-Linked
4
Figures :
Figure 1 : Première classification histopronostique des tumeurs cérébrales. ... 14
Figure 2 : Imagerie et histologie des tumeurs astrocytaires... 19
Figure 3 : Imagerie et histologie des tumeurs oligodendrocytaires. ... 21
Figure 4 : Imagerie et histologie des tumeurs mixtes ... 22
Figure 5 : Algorithme simplifié de la classification des gliomes selon l'OMS 2016 .. 24
Figure 6 : Implication des formes mutées d'IDH1 dans la gliomagenèse ... 26
Figure 7 : Propriétés des CSG ... 43
Figure 8 :Modèles proposés pour expliquer l’origine des CSG ... 44
Figure 9 : Facteurs intrinsèques et extrinsèques de régulation des CSG ... 46
Figure 10 : Plasticité métabolique des CSG ... 50
Figure 11 : Schéma du microenvironnement tumoral des CSG ... 53
Figure 12 : Voie de signalisation NOTCH ... 59
Figure 13 : Voie de signalisation WNT ... 61
Figure 14 : Voie de signalisation Hedgehog ... 62
Figure 15 : Structure des facteurs de transcription STAT ... 68
Figure 16 : Voie de signalisation STAT3... 70
Figure 17 : Implication de STAT3 dans les GBM ... 73
Figure 18 : Rôle de la signalisation Hippo dans la croissance des organes ... 80
Figure 19 : Voie de signalisation Hippo chez les mammifères ... 82
Figure 20 : Rôle de YAP1/TAZ dans les cellules souches cancéreuses ... 85
Figure 21 : Implication de YAP1/TAZ dans les tumeurs solides ... 88
5
différentiellement exprimés entre les sous-types moléculaires de gliomes... 174
Figure 25 : Identification de TEAD3 en tant que facteur de mauvais pronostic ... 175
Figure 26 : Rôle pronostique de TEAD3 dans une cohorte de 82 patients ... 177
Figure 27 :Etat d’activation de la voie de signalisation Hippo dans les CSG ... 179
Figure 28 :Etat d’activation des effecteurs YAP1 et TEAD3 dans les CSG ... 182
Figure 29 : Rôle biologique de TEAD3 dans les CSG ... 184
Tableaux :
Tableau 1 : Classification histopathologique de l'OMS 2007 des gliomes ... 16Tableau 2 : Rôles oncogéniques de YAP1/TAZ dans les tumeurs solides ... 90
Tableau 3 : Caractéristiques des 16 cultures de CSG ... 105
Tableau 4 : Liste des amorces utilisées en RT-qPCR ... 108
9
11
Les tumeurs cérébrales
I.
1. Epidémiologie
Les gliomes représentent 80% des tumeurs malignes du système nerveux central (SNC) avec une incidence d’environ 5 cas pour 100 000 personnes ce qui correspond à 27 000 nouveaux cas par an en Europe. L’incidence est plus élevée chez les hommes avec un taux de 5,4 contre 3,6 chez les femmes et augmente significativement avec l’âge passant de 0,9 chez les enfants à 12,1 chez les personnes de plus de 65 ans (Reni et al., 2017). Une augmentation de l’incidence chez les personnes âgées a également été observée ces dernières années, qui pourrait être due au vieillissement de la population et à l’amélioration des moyens de détection par imagerie (Crocetti et al., 2012).La forme la plus fréquente des gliomes est le glioblastome (GBM) qui représente plus de 50% des tumeurs malignes du SNC.
2. Facteurs de risques
L’étiologie des gliomes reste encore inconnue et aucun facteur de risque lié au mode de vie comme la consommation de tabac ou d’alcool, qui sont associés au développement de nombreux cancers, n’a été clairement établi pour ces tumeurs du SNC. Les radiations ionisantes représentent pour le moment le seul facteur de risque avéré pour les tumeurs cérébrales (Wrensch et al., 2002). En effet, une étude sur une cohorte de 680 000 personnes, ayant subi un examen par tomodensitométrie dans leur enfance ou à l’adolescence, a montré une augmentation d’un facteur 2 du risque de développer une tumeur cérébrale. Ce risque augmenterait avec la dose d’irradiation surtout chez les enfants (Mathews et al., 2013).
12
Avec l’explosion de l’utilisation des téléphones mobiles depuis le début des années 2000, de nombreuses études se sont interrogées sur les risques associés aux champs électromagnétiques de radiofréquences émis par des téléphones portables. Certaines études ont montré que l’exposition à long terme, ainsi qu’un usage régulier des téléphones mobiles augmentaient le risque de gliomes (Baan et al., 2011). En 2011, l’organisation mondiale de la santé (OMS) a catégorisé les champs électromagnétiques de radiofréquences en tant qu’élément carcinogène « potentiel ».
Des formes familiales de tumeurs cérébrales ont été rapportées et représentent environ 5% des cas (Wrensch et al., 1997). Il est intéressant de noter que le risque de développer un gliome chez l’enfant est augmenté avec le développement de cancers chez les parents tels que le mélanome ou le cancer de l’endomètre (Hemminki et al., 2001). En revanche, aucun lien n’a été trouvé entre le tabagisme ou la consommation d’alcool des parents et le risque de survenue d’un gliome chez l’enfant (Bailey et al., 2017).
Certains syndromes héréditaires peuvent être associés à une augmentation du risque de développer des gliomes. Par exemple, 15 à 20% des patients atteints de neurofibromatose de type 1, une maladie autosomale dominante causée par la mutation du gène NF1 (Neurofibromine 1); sont susceptibles de développer des tumeurs astrocytaires. D’autres maladies telles que le syndrome de Cowden, la neurofibromatose de type 2, la sclérose tubéreuse de Bourneville, le syndrome de Turcot de type 2 ou encore le syndrome de Li-Fraumeni sont associées à une prédisposition accrue au développement de gliomes. Au niveau génétique, des mutations dans le gène codant pour la p53 ou une perte d’hétérozygotie du chromosome 19 sont associées à un risque élevé de développer un gliome (Reni et al., 2017).
13
3. Symptômes et diagnostic
Les manifestations cliniques des tumeurs cérébrales ne sont pas spécifiques et les symptômes varient en fonction de la taille, de la localisation, du degré de malignité de la tumeur et de l’œdème cérébral associé. Les symptômes les plus fréquents sont les crises d’épilepsie présentées par 25% des patients au moment du diagnostic et 20% de patients en plus les développent au cours de la maladie (Reni et al., 2017). Les autres symptômes sont des maux de tête, des troubles de la vision et de la parole ainsi que de la coordination et de l’équilibre, des pertes de mémoire et de connaissance. Dans la plupart des cas, la tumeur est diagnostiquée quelques mois après l’apparition des premiers signes cliniques.
Un examen par imagerie par résonance magnétique (IRM) est généralement réalisé afin de déceler la présence d’une masse tumorale après l’apparition de ces symptômes. En IRM, les GBM présentent typiquement une prise de contraste au gadolinium avec un centre nécrotique souvent entouré d’un œdème péri-lésionnel. Cette technique permet de visualiser la localisation tumorale et le nombre de foyers tumoraux afin d’évaluer son opérabilité. De plus, les techniques d’imagerie permettent d’obtenir des informations sur le métabolisme tumoral et peuvent aller jusqu’à la prédiction de la survie des patients (Smits and van den Bent, 2017). Law et al. ont montré par l’IRM de perfusion que le pronostic des patients atteints de tumeurs de bas grade avec un haut niveau de perfusion était moins favorable que ceux atteint d’une tumeur de haut grade mais faiblement perfusés (Law et al., 2008). Cependant, un examen histologique et des analyses moléculaires sont nécessaires en plus de la neuro-imagerie afin d’identifier plus précisément le type de la tumeur.
14
Classifications histologiques des gliomes
II.
1. Historique
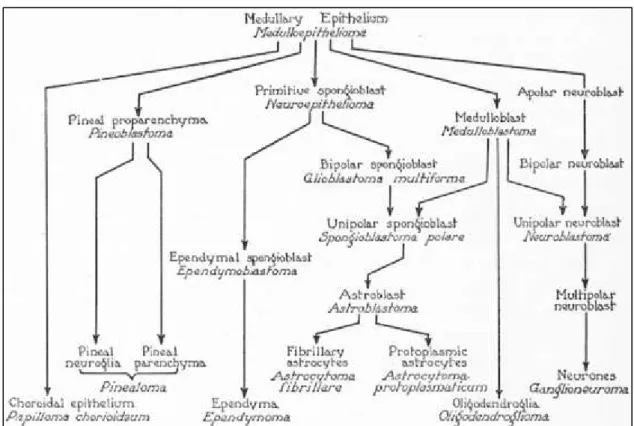
La première classification histopronostique des gliomes a été proposée par Percival Bailey et Harvey Cushing en 1927 (Figure 1) (Bailey and Cushing, 1927). Cette classification était basée sur la théorie histo-embryogénétique dans laquelle une valeur pronostique individuelle est attribuée à chacun des types ou sous-types tumoraux. Avant Bailey et Cushing, les classifications des tumeurs du SNC étaient uniquement descriptives. Ils se sont appuyés sur la théorie de Conheim qui postule que les tumeurs malignes naissent à partir des restes embryonnaires qui n’ont pas achevé leur différenciation et sur le principe de Mallory qui stipule que le comportement des tumeurs est directement fonction du type cellulaire prédominant (Nataf et al., 2005). Ainsi, la survie des patients serait d’autant plus courte que la tumeur reproduit un stade plus précoce dans l’échelle de maturation cellulaire.
Figure 1 : Première classification histopronostique des tumeurs cérébrales (d'après Bailey et Cushing, 1927).
15
En 1949, Kernohan introduit le concept d’anaplasie et à l’inverse des théories de Bailey et Cushing, considère que les tumeurs dérivent des cellules adultes par un processus de dédifférenciation (Kernohan et al., 1949). Il propose une classification en 4 grades de malignité croissante en fonction de la proportion des composantes tumorales différenciées ou indifférenciées dont il montre la corrélation avec la survie sur une cohorte de 161 patients.
Afin de trouver un consensus entre différents systèmes utilisés, l’OMS a proposé la première classification officielle des tumeurs cérébrales en 1979, révisée en 1993, 2000 et 2007 (Kleihues et al., 1993; Kleihues and Sobin Leslie, 2000; Louis et al., 2007 lch, 1979). Cette classification « histologique » est basée sur des critères cytologiques en fonction du type cellulaire prédominant et en 4 grades en fonction des signes de malignité tels que les atypies nucléaires, l’activité mitotique et la présence de nécrose. Les gliomes ont été ainsi répartis en 3 grands groupes : les astrocytomes, les oligodendrogliomes et les oligoastrocytomes ou tumeurs mixtes.
16
2. Classification de l’OMS 2007
Bien que la classification OMS 2007 est encore utilisée car elle ne nécessite pas d’analyses moléculaires, la version de 2016 représente le standard actuel de classification de tumeurs gliales (Tableau 1).
Type de tumeur Grade Différenciation Densité cellulaire Atypies nucléaires Activité mitotique Nécrose Prolifération vasculaire Survie médiane Oligodendrogliomes de bas grade II Haut degré de
différenciation Modérée Occasionnelles Occasionnelles Absente Absente
plus de 10 ans Oligodendrogliomes anaplasiques III Anaplasie focale ou dispersée
Elevée Présentes Présentes Présente Occasionnelle 5 ans Oligoastrocytomes
de bas grade II
Haut degré de
différenciation Modérée Occasionnelles Occasionnelles Absente Absente 6 ans Oligoastrocytomes
anaplasiques III
Anaplasie focale ou dispersée
Elevée Présentes Présentes Absente Occasionnelle 3 ans Astrocytomes diffus
de bas grade II
Haut degré de
différenciation Modérée Occasionnelles Occasionnelles Absente Absente 6-8 ans Astrocytomes anaplasiques III Anaplasie focale ou dispersée Augmentée diffusément ou focalement
Présentes Présentes Absente Absente 2-3 ans
Glioblastomes IV Faible Elevée Marquées Marquées Présente Présente 15 mois
17
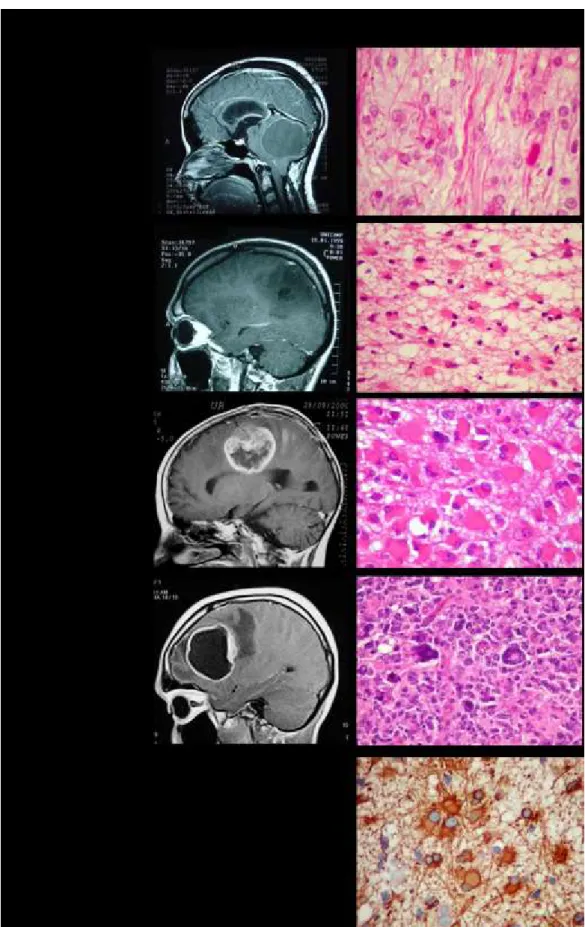
Les astrocytomes 1)
Les astrocytomes possèdent les caractéristiques morphologiques et immunohistochimiques des astrocytes : petites cellules de forme étoilée caractérisée par l’expression d’un marqueur cellulaire spécifique, la GFAP (Glial Fibrillary Acidic Protein) (Figure 2).
Les astrocytomes pilocytiques (grade I)
Ces tumeurs touchent principalement les enfants ainsi que les jeunes adultes et représentent environ 5% des tumeurs gliales. Elles peuvent se développer dans toutes les zones du cerveau, mais il existe une prédominance pour le cervelet. La résection chirurgicale complète permet généralement la guérison du patient. Cependant, dans de rares cas elles peuvent évoluer en gliomes malins.
Les astrocytomes diffus de bas grade (grade II)
Ce sont des tumeurs qui touchent les adultes dont le pic se situe aux alentours de 35 ans et représentent 5 à 10% des gliomes. Au niveau histologique, elles sont caractérisées par une légère augmentation de la cellularité, la présence occasionnelle d’atypies nucléaires et une absence ou une très faible présence de mitose. Cette tumeur est hypodense et ne présente pas de prise de contraste en imagerie. Les astrocytomes diffus de grade II évoluent généralement, dans environ 50% des cas, en tumeurs de grade III ou IV. La survie moyenne après l’intervention chirurgicale varie entre 6 et 8 ans.
18 Les astrocytomes anaplasiques (grade III)
Ces tumeurs représentent 10 à 30% des gliomes et évoluent rapidement en tumeurs de grade IV. L’âge médian auquel surviennent ces tumeurs est d’environ 40 ans et la survie après intervention chirurgicale est de 2 à 3 ans. Histologiquement, elles sont caractérisées par une plus forte densité cellulaire, la présence d’atypies nucléaires et une activité mitotique importante. En imagerie, la tumeur est hypodense et présente souvent une prise de contraste ou un œdème péri-tumoral.
Les glioblastomes (grade IV)
Les GBM représentent 50% des gliomes et surviennent principalement chez les personnes d’âge avancé. On distingue deux types de GBM : les GBM primaires dit de novo qui correspondent à la majorité des GBM et les GBM secondaires qui proviennent de l’évolution d’un gliome de bas grade. Histologiquement, en plus de la forte densité cellulaire, la présence d’atypies nucléaires et une activité mitotique importante, les GBM se caractérisent par la présence de nécrose entourée de cellules pseudopalissadiques, ainsi qu’une prolifération endothéliocapillaire. La survie médiane des patients est d’environ 15 mois après une exérèse chirurgicale et une radio-chimiothérapie concomitante. Cette tumeur se présente en imagerie sous forme d’une masse irrégulière avec prise de contraste annulaire et se développe généralement dans les hémisphères cérébraux au niveau des régions fronto-temporales et pariétales.
19
20
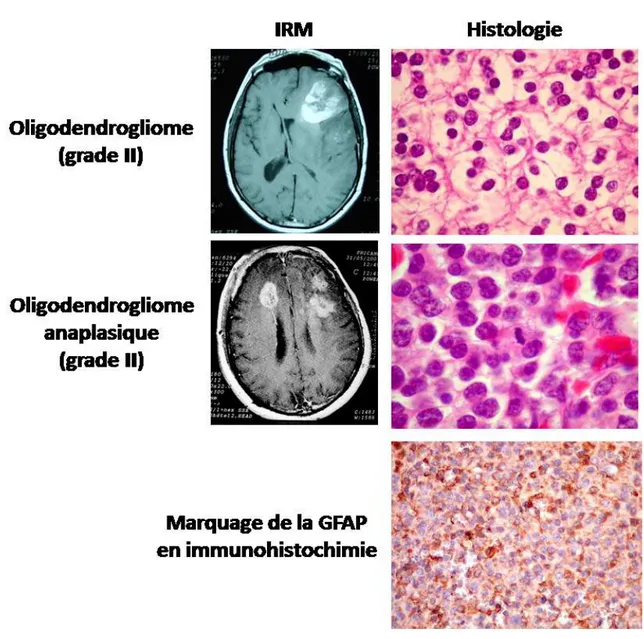
Les oligodendrogliomes 2)
Les oligodendrogliomes sont des tumeurs qui présentent une morphologie de type oligodendrocytaire (Figure 3). Elles constituent 5 à 25% des gliomes et présentent un meilleur pronostic que les astrocytomes.
Les oligodendrogliomes de bas grade (grade II)
Ce sont des tumeurs qui se développent généralement dans la substance blanche ou le cortex des hémisphères cérébraux et touchent les adultes entre 40 et 45 ans. La survie des patients peut atteindre plusieurs dizaines d’années après le diagnostic. Cette tumeur est caractérisée histologiquement par un aspect en « nid d’abeilles » et une morphologie cellulaire en « œuf au plat » car les cellules présentent un halo péri-nucléaire clair. La tumeur est hypodense et ne présente pas de prise de contraste en imagerie.
Les oligodendrogliomes anaplasiques (grade III)
Contrairement aux oligodendrogliomes de grade II, ces tumeurs présentent une prise de contraste qui peut être annulaire en imagerie. Elles se distinguent par la présence de cellules indifférenciées ainsi qu’une activité mitotique plus importante. Elles se développent chez les personnes entre 45 et 50 ans et sont de mauvais pronostic avec une survie médiane des patients d’environ 5 ans.
21
22
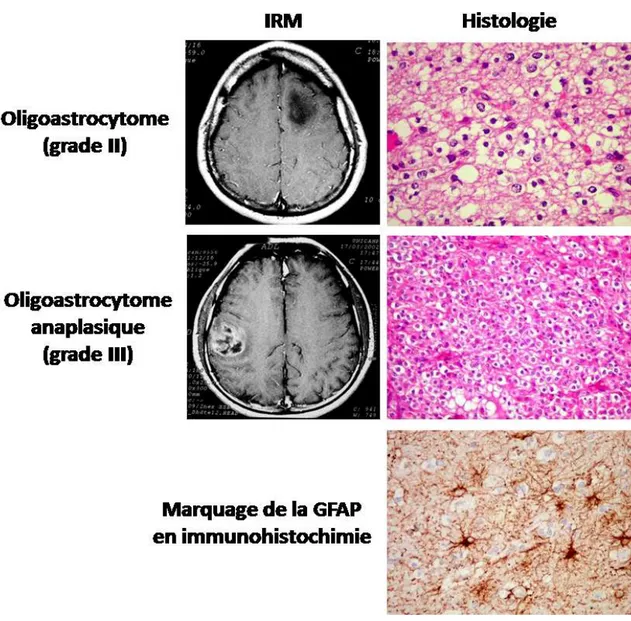
Les tumeurs mixtes 3)
Les oligoastrocytomes sont des tumeurs qui présentent à la fois des composantes cellulaires oligodendrocytaires et astrocytaires (Figure 4). Elles surviennent chez les personnes entre 35 et 45 ans et se localisent préférentiellement au niveau des hémisphères cérébraux. Elles peuvent être de bas grade (grade II) ou lorsqu’elles présentent des signes d’anaplasie, on parle d’oligoastrocytomes anaplasiques de grade III. La survie des patients est d’environ de 3 ans mais varie en fonction du grade.
23
3. Classification de l’hôpital St Anne
En 1997, le Pr. Daumas-Duport de l’hôpital Ste Anne a proposé une classification histo-radiologique des gliomes afin de pallier les problèmes de reproductibilité de la classification de l’OMS (Duport et al., 1997a; Daumas-Duport et al., 1997b). Cette classification est basée sur la structure spatiale ainsi que le mode de croissance des gliomes et distingue deux composantes tumorales : la forme infiltrante et la forme solide. La forme infiltrante est composée de cellules tumorales dans le parenchyme sain alors que la forme solide est uniquement constituée de cellules compactes et le passage de la forme infiltrante à solide s’accompagne d’une microangiogenèse. Deux grades de malignité ont été définis en fonction de l’hyperplasie endothéliale et de la prise de contraste en imagerie. Le grade A correspond à l’absence d’hyperplasie endothéliale et de prise de contraste alors que le grade B correspond à la présence d’hyperplasie et/ou de prise de contraste.
24
Classification de l’OMS 2016
III.
La classification de l’OMS des gliomes a été révisée en 2016 et intègre désormais des paramètres moléculaires permettant une meilleure stratification des patients et donc un diagnostic plus précis (Figure 5). Les mutations des gènes codant pour les enzymes Isocitrate déshydrogénases 1 et 2 (IDH1 et IDH2), ainsi que la codélétion du bras court du chromosome 1 et du bras long du chromosome 19 (1p19q) sont au cœur du diagnostic. D’autres mutations telles que les mutations dans les gènes TP53 (Tumor Protein 53) et ATRX (ATP-dependent helicase ATRX, X-linked helicase II) peuvent être des caractéristiques de définitions supplémentaires.
25
1. Statut IDH
Dans les cellules humaines, il existe trois types d’enzymes IDH qui interviennent dans le cycle de Krebs : IDH1, IDH2 et IDH3. IDH1 et 2 catalysent la décarboxylation oxydative de l’isocitrate en α-cétoglutarate (α-KG) en réduisant le NADP+ en NADPH, alors que l’enzyme IDH3 convertit le NAD+ en NADH. Plusieurs mutations dans les gènes IDH ont été rapportées. La mutation la plus fréquente représentant 90% de toutes les mutations d’IDH est la mutation c.395G>A d’IDH1 conduisant à la substitution d’une arginine par une histidine en position 132 (R132H) (Ohba and Hirose, 2018). Les autres formes mutantes de la protéine IDH1 sont R132C, R132S, R132G et R132L. Ils existent également des formes mutées de l’enzyme IDH2 généralement sur R172 qui sont beaucoup plus rares et présentes dans environ 3% des tumeurs. Les mutations d’IDH2 sont mutuellement exclusives avec les mutations d’IDH1. Initialement les mutations d’IDH1 ont été décrites en tant qu’inhibitrices de la fonction normale d’IDH1 dans la conversion de l’isocitrate en α-KG. Cependant, l’enzyme IDH1 tronquée garde sa fonction, mais produit une conversion incomplète d’isocitrate en 2-hydroxyglutarate (2-HG) et c’est ce nouveau produit de réaction qui pourrait être à l’origine de la gliomagenèse dans des tumeurs IDH1/2 mutées (Figure 6). En effet, il a été montré que l’accumulation de 2-HG induisait la production des dérivés réactifs de l'oxygène (ROS : « reactive oxygen species ») qui provoquent des dommages de l’ADN favorisant ainsi la transformation maligne des cellules (Zhang et al., 2013). Les mutations d’IDH1/2 sont ainsi présentes dans plus de 70% des astrocytomes et oligodendrogliomes de grade 2 et 3 ainsi que dans les GBM secondaires où elles sont associées à un meilleur pronostic chez les jeunes patients (Parsons et al., 2008; Yan et al., 2009). La
26
présence ou non de mutations dans IDH1/2 constitue le premier élément de discrimination des gliomes.
Figure 6 : Implication des formes mutées d'IDH1 dans la gliomagenèse (d’après Ohba et Hirose, 2018).
27
2. Codélétion 1p19q
Actuellement, la codélétion 1p19q constitue le deuxième élément moléculaire après le statut mutationnel d’IDH1/2 dans la classification OMS 2016. La codélétion 1p19q a été le premier marqueur moléculaire identifié dans les gliomes et a permis de distinguer les oligodendrogliomes des astrocytomes. En effet, la perte du 1p et du 19q a été observée dans 50% à 70% des oligodendrogliomes de grade II et III (Kraus et al., 1995). Par ailleurs, il a été constaté qu’au sein des oligodendrogliomes, environ un tiers des tumeurs de même morphologie étaient résistantes à la chimiothérapie sans qu’aucun marqueur clinique ou histopathologique ne puisse prédire la réponse au traitement. En 1998, Cairncross et al. ont montré que la codélétion 1p19q dans les oligodendrogliomes anaplasiques était associée à une meilleure survie et permettait de prédire la réponse à la chimiothérapie (Cairncross et al., 1998). De plus, des analyses multi-variées ont montré que cette codélétion était un marqueur indépendant du pronostic plus fort que des paramètres tels que l’âge des patients ou l’indice de Karnofsky. Finalement, depuis la classification OMS 2016, le groupe des tumeurs mixtes n’existe plus et la présence de la codélétion 1p19q constitue un élément clé dans la distinction entre les tumeurs oligodendrocytaires et astrocytaires. Les gliomes présentant des mutations d’IDH1/2 associées à la codélétion 1p19q correspondent aux tumeurs de meilleur pronostic avec la médiane de survie la plus longue.
28
3. Autres marqueurs moléculaires
Il est important de noter qu’il existe d’autres marqueurs moléculaires pris en compte dans la classification de l’OMS 2016 qui sont caractéristiques d’un groupe tumoral, mais ne sont pas essentiels au diagnostic. Il s’agit des mutations de TERT (Telomerase Reverse Transcriptase) et ATRX qui sont mutuellement exclusives, ainsi que celles de TP53. Les mutations de TERT sont associées au diagnostic des oligodendrogliomes en présence de la codélétion 1p19q ou des GBM en absence de la codélétion, alors que les mutations de TP53 et d’ATRX sont associées aux astrocytomes mutés sur IDH1/2.
En somme, la classification OMS 2016, en intégrant des paramètres moléculaires, a permis de palier la grande variabilité intra- et inter-observateurs dont souffrait la version précédente basée uniquement sur les caractéristiques histopathologiques en particulier pour les tumeurs de bas grade. On distingue donc globalement les GBM IDH1/2 sauvages (IDHwt) et les gliomes de bas grade IDHwt (GBG IDHwt) ou IDH1/2 mutés (GBG IDHmut), ces derniers pouvant être codélétés ou non sur 1p19q (GBG IDHmut codel ou GBG IDHmut non-codel).
29
Les glioblastomes
IV.
1. Caractéristiques des GBM
Les GBM sont la forme la plus fréquente et la plus agressive des gliomes. Ces tumeurs sont très hétérogènes à la fois sur le plan moléculaire et cellulaire. Elles sont caractérisées par une forte vascularisation et prolifération cellulaire, une anaplasie et la présence de plage de nécrose plus ou moins importante. On distingue les GBM primaires dit de novo non mutés sur IDH1/2 et les GBM secondaires provenant de l’évolution d’un gliome de bas grade présentant une mutation sur IDH1/2. Ces deux formes se distinguent également par différentes altérations moléculaires qui peuvent être mutuellement exclusives.
Les GBM primaires se distinguent par l’amplification et la surexpression de l’EGFR (Epidermal Growth Factor Receptor) retrouvées dans 40% de cas. L’EGFRvIII est un variant tronqué le plus fréquemment retrouvé correspondant à 50% des altérations de l’EGFR provenant de la délétion des exons 2 à 7 (Felsberg et al., 2017). Ce variant présente des altérations au niveau de son domaine extracellulaire résultant en une activation constitutive de l’EGFR et des voies de signalisation associées impliquées dans la prolifération, l’invasion et la survie cellulaire. Dans les GBM secondaires, les mutations les plus fréquentes après IDH1/2 touchent TP53 et sont retrouvées dans environ 65% des tumeurs (Ohgaki and Kleihues, 2013).
Au cours de ces vingt dernières années, de nombreuses études transcriptomiques à grande échelle ont permis de classer les GBM en fonction des signatures moléculaires.
30
2. Classification de Philipps
Cette première classification moléculaire est basée sur le profil d’expression d’une signature de 35 gènes et classe les GBM en 3 groupes : proneuraux, prolifératifs et mésenchymateux (Phillips et al., 2006). Les GBM proneuraux sont caractérisés par une expression des gènes impliqués dans le développement du SNC tels que DLL3 (Delta-like 3), OLIG2 (Oligodendrocyte transcription factor) ou BCAN (Brevican core protein) et sont dit de bon pronostic avec une survie médiane de 174,5 semaines. Le sous-type prolifératif est caractérisé par l’expression des gènes de la prolifération cellulaire comme PCNA (Proliferating Cell Nuclear Antigen) ou TOP2A (DNA topoisomerase 2-alpha) et est associé au mauvais pronostic avec une survie médiane de 60,5 semaines. Le type mésenchymateux est également de mauvais pronostic avec une survie médiane de 65 semaines et est associé à l’expression des gènes impliqués dans l’angiogenèse tels que CHI3L1/YKL40 (Chitinase-3-like protein 1), CD44 et VEGF (Vascular Endothelial Growth Factor). Un des points intéressant de cette étude et qu’elle montre que la plupart des GBM bascule vers un phénotype mésenchymateux lors de la récidive.
31
3. Classification de Verhaak
Une classification moléculaire plus récente des GBM a été proposée suite aux travaux du TCGA (The Cancer Genome Atlas). Cette signature est basée sur l’expression de 840 gènes et permet de distinguer 4 sous-types de tumeurs : classique, mésenchymateux, proneural et neural (Verhaak et al., 2010). Les GBM de type classique sont caractérisés par une amplification du chromosome 7 et par conséquent de l’EGFR associée à une perte du chromosome 10 et une absence de mutation de TP53. Dans ce groupe, on retrouve également une forte activation des voies de signalisation SHh (Sonic Hedegehog) et NOTCH avec surexpression des gènes associés comme SMO (Smoothened), GAS1 (Growth arrest-specific protein 1), GLI2 (Zinc finger protein GLI2), NOTCH3 et JAG1 (Jagged1). Les GBM mésenchymateux comme indique leur nom expriment fortement des marqueurs mésenchymateux tels que CHI3L1/YKL40, CD44, MERTK (Myeloid-Epithelial-Reproductive Tyrosine Kinase) et c-MET (Tyrosine-Protein Kinase Met). Ce sous-type est caractérisé par des altérations de la voie de signalisation PI3K/AKT (Phosphoinositide 3-kinase/ Protein kinase B) dues aux mutations de PTEN (Phosphatase and Tensin Homolog) et de la délétion 17q11 contenant le gène NF1. Ce sont des tumeurs présentant de fortes plages de nécrose et d’infiltrats inflammatoires du fait de la surexpression des gènes de la super famille des TNF (Tumor Necrosis Factor) et de la voie de signalisation NF-κB (Nuclear Factor kappa-light-chain-enhancer of activated B cells) tels que TRADD (Tumor necrosis factor receptor type 1-associated DEATH domain protein), RELB (RELB Proto-Oncogene, NF-κB Subunit ) et TNFRSF1A (Tumor Necrosis Factor Receptor Superfamily Member 1A). Les GBM de type proneural sont caractérisés avant tout par
32
l’amplification du PDGFRA (Platelet-Derived Growth Factor Receptor A) par altération du nombre de copie du locus 4q12 et la présence de mutations ponctuelles dans IDH1. Ce groupe présente également une forte expression des gènes impliqués dans le développement tels que NKX2.2 (NK2 homeobox 2), OLIG2, DLL3, SOX2 (Sex Determining Region Y-Box 2) et ERBB3 (Erb-B2 Receptor Tyrosine Kinase 3). Les tumeurs proneurales présentent beaucoup de similitudes avec les GBM secondaires. Enfin, les GBM de type neural sont associés à l’expression des marqueurs neuronaux tels que NEFL (Neurofilament Light), GABRA1 (Gamma-Aminobutyric Acid Type A Receptor Alpha1 Subunit), SYT1 (Synaptotagmin 1) et SLC12A5 (Solute Carrier Family 12 Member 5) mais ne présentent pas d’altérations génétiques spécifiques à ce groupe.
33
Les traitements des gliomes
V.
Les gliomes sont des tumeurs difficiles à traiter du fait de leur localisation et l’irréversibilité des lésions induites dans le cerveau. La stratégie thérapeutique est discutée lors de réunions de concertation pluridisciplinaire qui comprennent au moins un chirurgien, un oncologue et un radiothérapeute, ainsi qu’un pathologiste, un imageur et un médecin de soins palliatifs. La prise en charge du patient est composée d’un traitement symptomatique et d’un traitement étiologique. Le traitement symptomatique comporte généralement une corticothérapie antiœdémateuse, des antiépileptiques et des antithrombotiques alors que le traitement étiologique dépend des paramètres cliniques du patient. Dans le cas des GBM, le traitement standard consiste en une intervention chirurgicale associée à une radio-chimiothérapie concomitante (Stupp et al., 2005).
1. Chirurgie
L’intervention chirurgicale représente le traitement de première intention des gliomes lorsqu’elle est possible. Après cartographie de la tumeur par IRM, le chirurgien décide de l’opérabilité de la tumeur en fonction de sa localisation, du nombre de sites tumoraux et de l’état physiologique du patient, l’ensemble de ces paramètres permettant ainsi d’évaluer le rapport bénéfices-risques. L’exérèse chirurgicale doit être la plus large possible sans entrainer de risques fonctionnels majeurs pour le patient et afin de limiter la récidive de la tumeur. La chirurgie éveillée avec cartographie de stimulation constitue un progrès majeur dans la prise en charge des GBM et des gliomes de haut grade. Cependant, l’exérèse totale de la tumeur est rarement possible du fait de son caractère fortement infiltrant et doit donc être complétée par la radio-chimiothérapie.
34
2. Radiothérapie
La radiothérapie est utilisée dans le traitement des gliomes de haut grade, ainsi que de bas grade évolutifs. Elle est administrée soit en post-opératoire à visée curative, soit en palliatif lorsque la tumeur n’est pas opérable. Ce traitement consiste à détruire les cellules cancéreuses en leur infligeant des dommages dans l’ADN afin de réduire le volume tumoral ou de prévenir la récidive. Dans le cas des GBM, le protocole de radiothérapie comprend une dose totale de 60 Gy répartie en 30 fractions de 2 Gy par jour, 5 jours par semaine. Cependant, la radiothérapie comporte de nombreux effets indésirables et les GBM sont fortement radiorésistants.
3. Chimiothérapie
La chimiothérapie à base de témozolomide (TMZ), agent alkylant bloquant la réplication de l’ADN, est utilisée dans le traitement des gliomes de bas grade 1p19q codéletés et des GBM de façon adjudante et concomitante avec la radiothérapie. Cette association dans le cadre du protocole de prise en charge a permis d’augmenter la médiane de survie des patients à 14,6 mois contre 12,1 avec la radiothérapie seule (Stupp et al., 2005). Le TMZ est utilisé dans le protocole de Stupp à la dose de 75mg/m2 par jour pendant 6 semaines 1 heure avant la
radiothérapie suivis par le traitement adjuvant à la dose de 150-200mg/m2 par jour
35
4. Autres traitements
Les nitrosourées 1)
Les nitrosourées telles que la carmustine (BCNU) et la lomustine (CCNU) sont des agents alkylants très largement utilisés dans la chimiothérapie de première intention des gliomes dans les années 1970 et 1980. Ces molécules sont très lipophiles ce qui facilite le passage de la barrière hémato-encéphalique (BHE). Cependant ces composés sont extrêmement toxiques ce qui provoquent de nombreux effets secondaires rendant leur utilisation limitée en clinique. Actuellement, les nitrosourées peuvent être utilisées en seconde ligne pour le traitement des GBM récidivants (Brandes et al., 2016).
Le traitement ciblant les récepteurs tyrosine-kinases (RTK) 2)
Le récepteur à l’EGF constitue une cible de choix dans les GBM dans lesquels il est retrouvé fréquemment muté ou surexprimé. Les inhibiteurs des récepteurs à activité tyrosine kinase (ITK) tels que l’erlotinib ou le gefitinib ont montré des premiers résultats prometteurs dans des études précliniques en inhibant la croissance tumorale et en augmentant la survie. Cependant, ces effets n’ont pas été concluants après les études cliniques du fait de la faible perméabilité de la BHE à ces molécules limitant par conséquent leur utilisation dans le traitement des GBM (An et al., 2018). Une thérapie ciblée visant l’EGFRvIII avec l’ABT-414 qui est un agent constitué d’un anticorps anti-EGFR conjugué avec un inhibiteur de la tubuline, la monométhyl-auristatine F, a également été proposée avec des effets encourageants dans les phases précliniques. Le développement clinique de l'ABT-414 se poursuit avec des essais cliniques randomisés de phase II/III. D’autres
36
récepteurs tyrosines kinases tels que FGFR (Fibroblast Growth Factor Receptor) et PDGFR sont aussi étudiés en tant que cibles thérapeutiques dans les GBM (Touat et al., 2017).
Le traitement anti-angiogénique 3)
Les GBM sont des néoplasmes infiltrants fortement angiogéniques. En effet, la tumeur doit générer de nouveaux vaisseaux afin d’être alimentée en oxygène et nutriments nécessaires à sa croissance. Les cellules tumorales sécrètent de nombreux facteurs proangiogéniques et vasculogéniques tels que le VEGF afin de recruter des cellules endothéliales in situ ou des précurseurs circulants. Dans le but de cibler cette angiogenèse tumorale, le bevacizumab, un anticorps monoclonal humanisé, a été proposé en tant que médicament potentiel (Avastin®) dans le traitement des GBM (Cohen et al., 2009). Bien que le bevacizumab permette d’atténuer la taille de la tumeur, il a été montré que son utilisation favorisait le passage d’une angiogenèse profuse à une invasion large du parenchyme cérébral sain (de Groot et al., 2010). De plus, une étude comparant l’effet du bevacizumab associé au protocole de Stupp par rapport au protocole de référence seul a montré une augmentation de la survie sans progression (6,2 mois contre 10,6) sans toutefois améliorer la survie globale des patients (16,8 mois contre 16,7) (Chinot et al., 2014). Enfin, il semblerait que le traitement avec le bevacizumab altère la qualité de vie des patients en entrainant un déclin plus rapide des fonctions neurocognitives (Gilbert et al., 2014).
37
Immunothérapies 4)
L'immunothérapie a suscité un intérêt considérable au cours des dernières années en tant que piste pour le traitement des GBM. Le ciblage thérapeutique du point de contrôle immunitaire PD1 (Programmed Cell Death 1) et de son ligand PD-L1, ainsi que de la CTLA-4 (Cytotoxic T-Lymphocyte-Associated molecules-4) avec des anticorps spécifiques a été associé à un bénéfice clinique significatif dans les mélanomes et le cancer bronchique non à petites cellules (Garon et al., 2015; Robert et al., 2015). Dans les GBM, PD-L1 est exprimé chez certains patients et des études précliniques ont justifié l'évaluation des bloqueurs de points de contrôle immunitaires ICB (Immune Checkpoint Blockers) (Touat et al., 2017). Plusieurs essais cliniques évaluant les ICB sont en cours, notamment des essais randomisés de phase III sur le nivolumab ciblant le PD1 (NCT02617589, NCT02667587, NCT02017717). D'autres approches basées sur les cellules immunitaires modifiées, telles que les lymphocytes NK (Natural Killer), réorientées vers des antigènes tumoraux spécifiques comme l’EGFRvIII, ont démontré une efficacité antitumorale prometteuse dans des modèles animaux et sont actuellement évaluées dans plusieurs essais de phase I/II (NCT01109095, NCT02442297, NCT02664363, NCT01454596) (Johnson et al., 2015).
39
Chapitre II
Les Cellules Souches de
Glioblastomes
41
Les cellules souches cancéreuses
I.
1. Découverte
Les cellules souches cancéreuses (CSC) ont été initialement isolées en 1997 dans la leucémie myéloïde aigue où ces cellules cancéreuses partagent les mêmes marqueurs CD34+ et CD38- que les cellules souches hématopoïétiques (Bonnet and
Dick, 1997). Par la suite, elles ont été isolées dans de nombreux cancers où elles représentent une faible population cellulaire capable d’initier et de régénérer la tumeur. Les cellules souches de glioblastomes (CSG) ont été mises en évidence par Ignatova et al. en les isolant et cultivant sous forme de neurosphères à partir de prélèvements issus de patients (Ignatova et al., 2002). Singh et al. ont montré par la suite la capacité tumorigénique des CSG, après les avoir triées sur la base de l’expression du marqueur CD133 (Prominine-1) et les avoir greffées chez les souris nude (Singh et al., 2003 ; Singh et al., 2004). Dans un même temps, deux équipes ont isolé des CSG par la méthode dite du « neurosphère assay » permettant d’analyser les caractéristiques fondamentales des cellules souches comme la prolifération, l’autorenouvellement et la multipotence (Galli et al., 2004 ; Yuan et al., 2004). Les CSG partagent donc des caractéristiques communes avec les cellules souches neurales « normales » (CSN). Cependant, contrairement à ces dernières les CSG sont capables de former des tumeurs phénotypiquement proches de la tumeur d’origine lorsqu’elles sont greffées chez les souris immunodéprimées. Pour être considérées comme souches, les cellules tumorales doivent donc remplir ces caractéristiques (Vescovi et al., 2006). L’origine exacte des CSG n’est pas encore clairement établie mais les théories actuelles postulent qu’elles pourraient provenir
42
de la transformation maligne d’une CSN ou d’un précurseur glial voir de la dédifférenciation d’une cellule gliale mature.
2. Propriétés
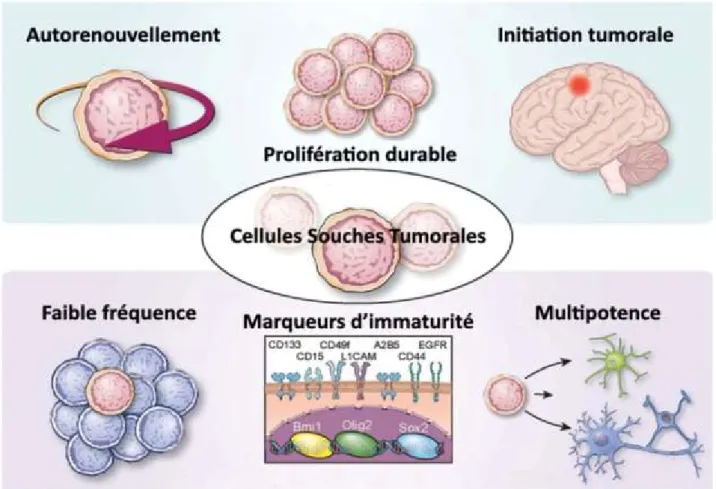
Les CSG ont la capacité de croitre sous forme de neurosphères dans les conditions de culture établies pour les CSN, dans un milieu dépourvu de sérum et supplémenté en EGF (Epidermal Growth Factor) et bFGF (basic Fibroblast Growth Factor) empêchant ainsi leur différentiation. Après dissociation des neurosphères, les CSG sont capables de reformer des sphères secondaires par prolifération clonale correspondant à leur capacité d’autorenouvellement. Comme les CSN, les CSG expriment des marqueurs souches tels que CD133, L1CAM (L1 Cell Adhesion Molecule), CD15 et la NESTINE. Le maintien du phénotype souche et de la capacité d’autorenouvellement ainsi que de la prolifération et la survie des CSG sont régulés par des facteurs de transcription tels qu’OLIG2, SOX2, OCT4 (Octamer-binding transcription factor 4), NANOG et c-MYC. En présence de sérum, les CSG sont capables de se différencier dans les 3 lignages du SNC, dont elles vont exprimer des marqueurs spécifiques : la GFAP pour les astrocytes, la β-Tubuline III pour les neurones et GALC (Galactosylcéramide) pour les oligodendrocytes. Lorsque les CSG sont greffées chez des souris immunodéficientes, elles sont non seulement capables de générer une tumeur mais cette capacité tumorigénique est maintenue lorsque des greffes sériées sont réalisées. (Figure 7) (Galli et al., 2004; Singh et al., 2004; Vescovi et al., 2006; Yuan et al., 2004).
43
3. Origines
Plusieurs théories ont été proposées pour expliquer l’existence de « cellules souches cancéreuses » au sein des tumeurs (Figure 8). La première théorie de l’origine des CSG est basée sur le modèle hiérarchique stipulant que la transformation prend place dans une cellule souche qui serait responsable de l’initiation et de la rechute tumorale. L’hétérogénéité de la tumeur proviendrait de cette cellule souche transformée pouvant donner naissance à la fois à d’autres cellules souches par autorenouvellement ou bien à des cellules engagées dans la
44
différenciation par un mécanisme de division asymétrique. Ces cellules filles vont à leur tour accumuler des mutations privées générant de l’hétérogénéité au sein de la tumeur. Le deuxième modèle proposé a été le modèle de l’évolution clonale reposant sur l’idée que toutes les cellules de la tumeur ont pu être transformées, qu’il s’agisse de cellules souches ou de cellules différenciées, et pourront acquérir une hétérogénéité mutationnelle. Actuellement, c’est un modèle hybride qui est proposé selon lequel les CSG peuvent provenir de la dédifférenciation et de la transdifférenciation des différents précurseurs cellulaires en réponse aux signaux intra et extracellulaires, et sont à l’origine de la récidive et de l’hétérogénéité tumorale (Thomas and Yu, 2017).
Figure 8 : Modèles proposés pour expliquer l’origine des CSG : modèle hiérarchique (A), modèle clonal (B) et modèle hybride (C) (d’après Thomas et Yu, 2017).
45
Hétérogénéité des CSG
II.
La plupart des marqueurs utilisés pour identifier les cellules souches cancéreuses sont basés sur ceux connus des cellules souches normales, tels que les différents facteurs de transcription et protéines structurales essentiels comme SOX2, NANOG, OLIG2, c-MYC, MUSASHI1, BMI1, NESTINE et ID1 (Inhibitor Of DNA Binding 1) (Anido et al., 2010; Ben-Porath et al., 2008; Hemmati et al., 2003; Kim et al., 2010; Ligon et al., 2007; Tunici et al., 2004). De nombreux marqueurs membranaires ont été proposés afin d’isoler rapidement les CSG par cytométrie en flux tels que CD133, CD15, CD44, intégrine α6, L1CAM ou encore A2B5 (Bao et al., 2008; Hemmati et al., 2003; Lathia et al., 2010; Liu et al., 2006; Ogden et al., 2008; Son et al., 2009). Ils interviennent dans l’interaction entre les cellules et leur microenvironnement et nécessitent une utilisation rapide car ils sont rapidement dégradés après la mise en culture. Le marqueur de surface exprimé par les CSN, CD133 a été le premier utilisé afin d’isoler les CSG car il est enrichi dans les cellules présentant une forte capacité d’autorenouvellement, de prolifération et de différentiation (Singh et al., 2003). Cependant, l’expression de CD133 et de son épitope de surface AC133 doit être utilisée avec précaution pour l’isolement des CSG. En effet, il a été montré que la différentiation cellulaire induisait une diminution de l’épitope AC133, alors que l’expression de CD133 restait inchangée (Kemper et al., 2010). De plus, des cellules initiatrices de tumeurs n’exprimant pas CD133 ont été isolées montrant ainsi que ce dernier ne peut pas être considéré en tant que marqueur universel des CSG (Beier et al., 2007). Les autres marqueurs proposés permettent de marquer une grande partie de la population de CSG, mais présentent au même titre que CD133 un nombre important de faux positifs confirmant que
46
Figure 9 : Facteurs intrinsèques et extrinsèques de régulation des CSG (d'après Lathia et al., 2015).
plusieurs méthodes complémentaires validant les caractéristiques des CSG sont nécessaires.
Facteurs de régulation des CSG
III.
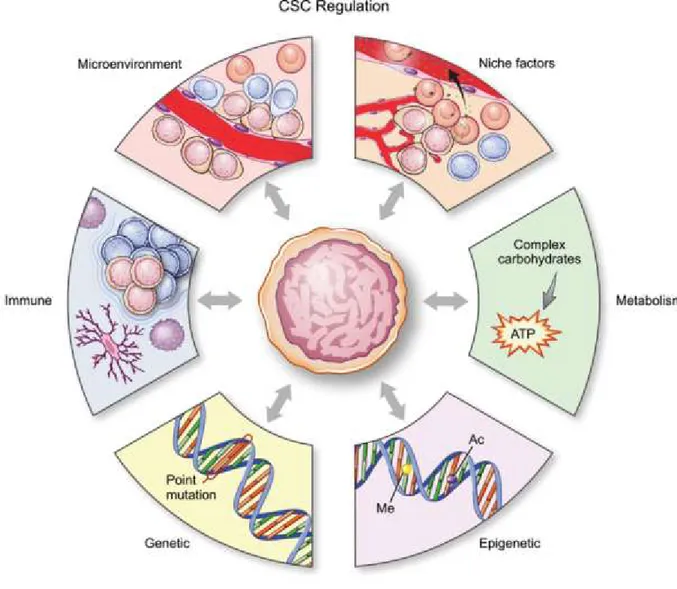
Les CSG sont régulées par 6 mécanismes majeurs répartis d’une part en facteurs intrinsèques tels que le profil génétique, épigénétique et métabolique, et d’autre part en facteurs extrinsèques comme le microenvironnement cellulaire, les facteurs sécrétés au sein de la niche et la réponse immune (Figure 9) (Lathia et al., 2015).
47
1. Les facteurs intrinsèques
Facteurs génétiques et épigénétiques 1)
Avec les avancées récentes dans le domaine de la génomique, beaucoup de mutations génétiques et de variants structuraux ont été décrits dans les GBM (Brennan et al., 2013; Cancer Genome Atlas Research, 2008). Les altérations les plus fréquentes touchent les gènes EGFR, IDH1, PDGFRA, MDM2, PI3KCA et le promoteur de TERT. De plus, les mutations ou les délétions induisant des pertes de fonctions de gènes suppresseurs de tumeurs tels que PTEN, TP53, CDKN2A (Cyclin Dependent Kinase Inhibitor 2A), NF1, ATRX ou RB1 (Retinoblastoma-associated protein) sont fréquemment retrouvées dans les GBM. L’ensemble de ces altérations montre une très forte hétérogénéité génétique des GBM ce qui souligne la complexité de l’évolution et de la diversité clonale des CSG qui pourraient être à l’origine de la tumeur (Phillips et al., 2006; Verhaak et al., 2010).
Les CSG sont régulées au niveau transcriptionnel par des facteurs de transcription variés en association avec les éléments de remodelage de la chromatine. Le facteur de transcription c-MYC semble être un élément clé de la régulation des CSG en contrôlant les mécanismes de prolifération et de survie cellulaire (Chen et al., 2012; Fang et al., 2014; Wang et al., 2008; Wurdak et al., 2010; Zheng et al., 2008). D’autres facteurs de transcription indispensables ont été identifiés incluant STAT3 (Signal Transducer And Activator Of Transcription 3), SOX2, FOXM1 (Forkhead Box M1), GLI1, ASCL1(Achaete-Scute Family BHLH Transcription Factor 1), ZFX (Zinc Finger Protein X-Linked), NANOG et ZFHX4 (Zinc Finger Homeobox 4) et nécessitent un recrutement des facteurs de remodelage de la
48
chromatine afin de maintenir les cellules cancéreuses dans un état souche (Chudnovsky et al., 2014; Clement et al., 2007; Fang et al., 2014; Gangemi et al., 2009; Joshi et al., 2013; Rheinbay et al., 2013; Sherry et al., 2009; Verginelli et al., 2013; Zbinden et al., 2010). En 2014, 4 facteurs de transcription clés des GBM proneuraux, POU3F2 (POU Class 3 Homeobox 2), SOX2, SALL2 (Spalt Like Transcription Factor 2) et OLIG2 ont été identifiés par cartographie de l’épigénome de la chromatine et sont capables de reprogrammer les cellules tumorales différenciées en CSG (Suva et al., 2014).
En plus des facteurs de transcription, les régulateurs de la structure du nucléosome participent également au maintien du phénotype souche des CSG. La protéine MLL1 (Mixed Lineage Leukemia 1) par l’intermédiaire de l’activation du réseau de gènes de la famille HOX, ainsi que la méthylase E H2 de l’histone H3K27 ont été montrées comme nécessaires au maintien du phénotype des CSG (Gallo et al., 2013; Heddleston et al., 2012; Kim et al., 2013b).