Université de Sherbrooke
Étude des déterminants de l’obésité infantile : Rôle de l'épigénétique dans la programmation fœtale de l’adiposité
Par
Valérie Gagné-Ouellet Programme de biochimie
Thèse présentée à la Faculté de médecine et des sciences de la santé en vue de l’obtention du grade de Philosophiae Doctor (Ph.D.)
en biochimie
Sherbrooke, Québec, Canada Janvier 2020
Membres du jury d’évaluation Michelle Scott – Présidente du jury Luigi Bouchard – Directeur de recherche
Jean-Luc Ardilouze – Évaluateur externe au programme
Serge McGraw – Évaluateur externe à l’Université (département d’obstétrique et gynécologie, Université de Montréal)
À ma famille, mes amis, mon conjoint et les générations qui nous suivront, puissiez-vous suivre vos rêves et chérir vos passions, avec courage, persévérance et beaucoup d’audace.
“Time present and time past are both perhaps present in time future, and time future contained in time past.” T.S. Eliot, Four Quartets
RÉSUMÉ
Étude des déterminants de l’obésité infantile: rôle de l'épigénétique dans la programmation fœtale de l’adiposité
Par
Valérie Gagné-Ouellet Programme de biochimie
Thèse présentée à la Faculté de médecine et des sciences de la santé en vue de l’obtention du diplôme de Philosophiae Doctor (Ph.D.) en biochimie, Faculté de médecine et des sciences de la santé, Université de Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4
L’obésité est aujourd’hui considérée comme un problème de santé majeure, promu au rang d’épidémie mondiale. Les prédictions de prévalence de l’obésité infantile sont encore plus qu’inquiétantes, rendant la découverte des facteurs étiologiques et mécanistiques essentielle à la mise sur pied de stratégies préventives et curatives. Bien que la sédentarité et l’alimentation, deux facteurs obésogènes, aient longtemps été nommées principales responsables de l’apparition de ce désordre métabolique, il est aujourd’hui estimé que la santé maternelle pourrait agir à titre de contributeur substantiel. L’exposition fœtale à l’hyperglycémie maternelle en cours de grossesse compliquée par un diabète gestationnel est désormais acceptée comme un facteur de risque d’obésité infantile, mais les mécanismes moléculaires sous-jacents doivent encore être identifiés. La méthylation de l’ADN, une modification épigénétique permettant de réguler l’expression génique, pourrait être impliquée dans la programmation fœtale de l’obésité infantile en réponse à une exposition à l’hyperglycémie maternelle. Le but de cette thèse est donc d’identifier les déterminants épigénétiques impliqués dans la programmation fœtale de l’obésité infantile reliée au DG. Par conséquent, on a montré que que des approches par gènes candidats et panépigénomiques ont été réalisées. En premier lieu, des différences de méthylation dans les gènes candidats de la lipoprotéine lipase et de la leptine sont associées à l’adiposité dans l’enfance. Ces changements de méthylation de l’ADN pourraient être induits par des expositions à l’hyperglycémie maternelle durant la grossesse. En second lieu, le méthylome placentaire a été analysé à l’aide de micropuces de méthylation de l’ADN chez les 259 participants de la cohorte Gen3G. Ces analyses ont permis d’identifier des sites de méthylation de l’ADN et des régions génomiques différentiellement méthylées associées à l’adiposité dans l’enfance. Les niveaux de méthylation de l’ADN des CpG a également été associée à l’expression génique de FAM3C et TFAP2E, deux acteurs potentiels de la différenciation des cellules adipeuses au cours du développement fœtal et/ou dans l’enfance. Ces résultats suggèrent que l’obésité infantile pourrait tirer, du moins en partie, son origine dans la réponse épigénétique à certaines perturbations métaboliques comme le diabète gestationnel et qui sont susceptibles d’induire l’accrétion excessive de masse adipeuse dans l’enfance.
SUMMARY
Identification of risk factors of childhood obesity: Role of epigenetic in the fœtal programming of adiposity
By
Valérie Gagné-Ouellet Biochemistry Program
Thesis presented at the Faculty of medicine and health sciences for the obtention of Doctor degree diploma Philosophiae Doctor (Ph.D.) in biochemistry, Faculty of medicine and
health sciences, Université de Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4 Obesity is now considered an overwhelming health problem, particularly since it has been promoted to the stage of a global epidemic, with increasing prevalence in all age groups. Predictions of childhood obesity prevalence rates are more than worrying, making the discovery of causal etiological and mechanistic factors essential to developing preventive and curative strategies. Although sedentary lifestyle and obesogenic diet have long been identified as the main causes of this metabolic disorder, it is now known that maternal health could act as a substantial contributor. Fœtal exposure to maternal hyperglycemia during pregnancy complicated by gestational diabetes mellitus is now recognized as a risk factor for childhood obesity, but the underlying molecular mechanisms are yet to be identified. DNA methylation, an epigenetic modification known to regulate gene expression, may be involved in the fœtal programming of childhood obesity in response to exposure to maternal hyperglycemia. The aim of this thesis was therefore to identify the epigenetic determinants involved in the fœtal programming of childhood obesity in response to gestational diabetes. Accordingly, candidate gene and panepigenomic approaches have been carried out. First, placental epimutations with functional impacts in the candidate lipoprotein lipase and leptin genes have been associated with adiposity in childhood. These DNA methylation differences could be induced by early exposures to abnormal maternal blood glucose levels during pregnancy and these results were confirmed by mediation analyses in these two separate studies. Second, placental methyloma was analyzed for 259 participants in the Gen3G cohort. These analyses identified childhood adiposity-associated epimutations for 2 CpG, with confirmed results in an independent cohort (n=187). DNA methylation of these epimutations has also been associated with the transcriptional activity of the FAM3C and TFAP2E genes, potential actors in the differentiation of adipose cells commitment during fœtal development or in childhood. Together, these results demonstrate that childhood obesity may be from fœtal origin, at least in part, and that metabolic disturbances such as the gestational diabetes are likely to modulate body fat accretion in childhood via alterations in the DNA methylation profile.
TABLE DES MATIÈRES
Résumé ... iv
Summary ... v
Table des matières ... vi
Liste des figures ... ix
Liste des tableaux ... xi
Liste des abréviations ... xii
Chapitre 1 - Introduction ... 1
1.1. Le portrait de l’obésité ... 1
1.1.1. La physiopathologie ... 3
1.1.2. L’étiologie et les facteurs de risque... 5
1.1.2.1. Les facteurs intrinsèques ... 6
1.1.2.1.1. L’obésité monogénique ... 7
1.1.2.1.2. L’obésité syndromique ... 8
1.1.2.1.3. L’obésité polygénique ... 10
1.1.2.2. Les facteurs extrinsèques ... 14
1.1.2.2.1. L’obésité infantile ... 16
1.2. Le portrait de l’obésité infantile ... 18
1.2.1. Le rapport de commission pour mettre fin à l’obésité infantile ... 18
1.1. Le développement prénatal ... 20
1.1.1. La phase pré-embryonnaire... 21
1.1.2. La phase embryonnaire ... 21
1.1.3. Le développement fœtal ... 23
1.2. Les origines développementales de la santé ... 24
1.2.1. Le portrait et la prévalence du diabète gestationnel ... 26
1.2.1.1. L’homéostasie glucidique durant la grossesse ... 26
1.2.1.2. L’insulino-résistance dans le diabète gestationnel... 28
1.2.1.3. Les conséquences maternelles du diabète gestationnel ... 31
1.2.1.4. Les conséquences du diabète gestationnel pour l’enfant : l’hypothèse de Pedersen 32 1.2.2. Les modifications épigénétiques comme acteurs du DOHaD ... 35
1.2.2.2. La transmissibilité des modifications épigénétiques ... 38
1.2.2.3. La plasticité de l’épigénome ... 39
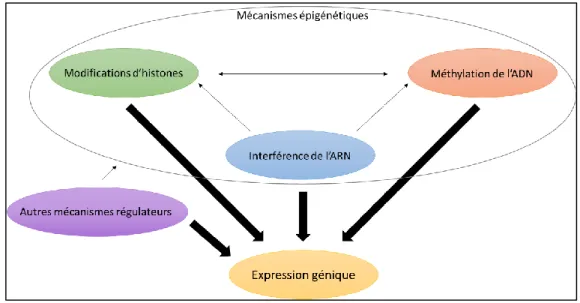
1.2.2.4. La régulation de l’expression génique ... 40
1.2.2.5. Les modifications d’histones ... 41
1.2.2.6. Les ARN non-codants ... 43
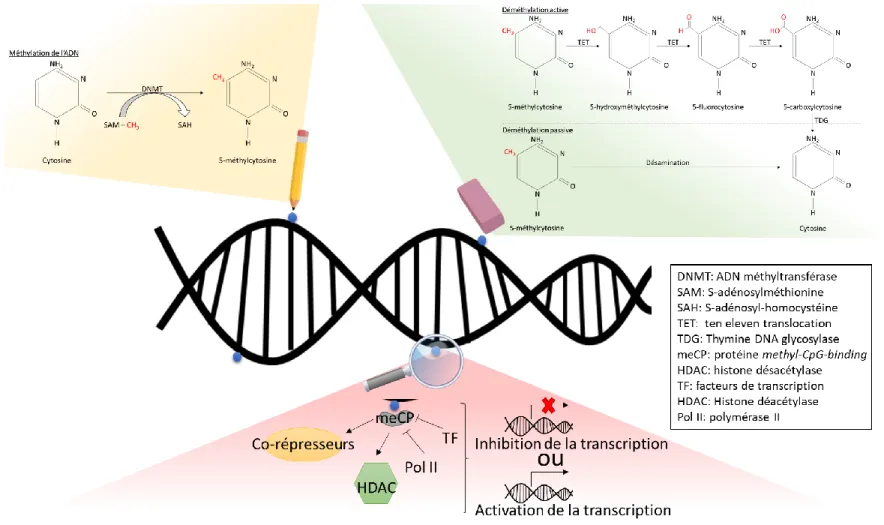
1.2.3. La méthylation de l’ADN ... 46
1.2.3.1. Les partenaires d’interactions de la méthylation de l’ADN ... 46
1.2.3.1.1. L’établissement du profile de méthylation de l’ADN ... 48
1.2.3.1.2. Le processus de déméthylation de l’ADN ... 49
1.2.3.1.3. La régulation de l’expression des gènes par la méthylation de l’ADN ... 51
1.2.3.2. Le rôle de la méthylation de l’ADN dans le développement prénatal ... 53
1.2.3.2.1. La reprogrammation et la différenciation cellulaire ... 53
1.2.3.2.2. L’empreinte parentale ... 56
1.2.3.2.3. L’inactivation du chromosome X ... 57
1.2.3.2.4. L’inactivation des retrotransposons ... 58
1.3. L’étude du méthylome dans un contexte de programmation fœtale de la santé métabolique ... 59
1.3.1. Les modèles animaux ... 60
1.3.2. Les sujets humains ... 62
1.3.2.1. L’approche par gène candidat ... 63
1.3.2.2. L’approche panépigénomique ... 65
1.4. L’étude du méthylome placentaire dans la programmation fœtale de l’obésité infantile ... 67 1.5. Problématique ... 70 1.5.1. Hypothèse ... 71 1.5.2. Objectifs ... 71 Chapitre 2 - Article 1 ... 72 Chapitre 3 - Article 2 ... 112
Table 3.1 Clinical characteristics of the mother-child dyads from the Gen3G ... 125
Chapitre 4 - Article 3 ... 147
Chapitre 5 - Discussion ... 194
5.1.1 L’impact d’un dérèglement épigénétique de la LPL placentaire... 195
5.1.2 Les répercussions d’une perturbation des niveaux de leptine circulants ... 197
5.1.3 L’identification de nouveaux déterminants épigénétiques à l’aide de l’approche panépigénomique ... 201
5.1.3.1 Le locus 7q31.3 ... 202
5.1.3.1.1 FAM3C... 203
5.1.3.2 TFAP2E ... 205
5.1.3.3 Les régions épigénomiques ... 207
5.1.3.4 Les voies biologiques enrichies ... 208
5.1 L’origine fœtale de l’obésité infantile ... 210
5.2 Potentiel clinique des épivariants identifiés ... 213
5.3 L’impact d’une exposition à l’hyperglycémie maternelle pour la santé de l’enfant .. 216
5.4 Les forces et les limitations de notre étude ... 219
5.5 La réplication des résultats dans une cohorte indépendante ... 220
5.5.1 La stabilité et la tissu-spécificité de la méthylation de l’ADN ... 221
5.5.2 Le choix des facteurs biologiques potentiellement confondants ... 222
5.5.3 Les interactions entre la génétique et l’épigénétique ... 224
5.6 Les avenues potentielles de ce projet ... 225
Chapitre 6 – Conclusions ... 229
Remerciements ... 230
Liste des références... 232
Annexe 1... 275 Annexe 2... 276 Annexe 3... 277 Annexe 4... 278 Annexe 5... 279 Annexe 6... 280 Annexe 7... 281 Annexe 8... 282 Annexe 9... 283 Annexe 10 ... 284 Annexe 11 ... 285
LISTE DES FIGURES
Figure 1.1 Chronologie du développement prénatal en cours de grossesse. ... 20 Figure 1.2 Origine embryonnaire des tissus et organes. ... 23 Figure 1. 3 Variations des niveaux d'insuline en cours de grossesse normoglycémique et avec
diabète gestationnel. ... 28 Figure 1. 4 Mécanismes physiologiques de régulation du glucose lors de grossesses avec ou
sans diabète gestationnel. ... 31 Figure 1. 5 Implication des mécanismes épigénétiques dans la régulation de l’expression
génique. ... 41 Figure 1. 6 Plasticité de la méthylation de l’ADN et régulation de l’expression géniques. . 47 Figure 2.1 Schematic representation of the LPL gene and localization of the three
epigenotyped regions. ... 82 Figure 2. 2 Comparison of anthropometric profiles in GDM and NGT offspring with WHO
recommendations. ... 85 Figure 2. 3 Associations between birth weight and body composition at age 5. ... 86 Figure 2. 4 Associations between placental DNA methylation levels at LPL CpG dinucleotide
3.4 and children's body composition... 88 Figure 3. 1 Schematic representation of the LEP gene and localization of the epigenotyped
CpG sites. ... 121 Figure 3.2 Associations between placental LEP DNA methylation (DNAm) and childhood
adiposity. ... 129 Figure 3.3 Mediation analysis between maternal fasting glucose at 2nd trimester of pregnancy,
placental LEP DNA methylation (DNAm) variations, and neonatal leptin concentrations. ... 131 Figure 4.1 Epigenome-Wide single-site associations between placental DNAm levels and
childhood adiposity. ... 164 Figure 4. 2 Association between DNAm levels at cg22593959 (A) and cg22436429 (B) and
Figure 4.3 Regional association plot and correlation heatmap of CpGs located near cg22593959 from the CpG-by-CpG analysis. ... 166 Figure 4.4 Regional plot and correlation heatmap of CpGs located near cg22436429 from the
CpG-by-CpG analysis. ... 167 Figure 4.5 Regional plot and correlation heatmap of CpGs located in the FMN1 gene locus
from the regional analysis. ... 170 Figure 4. 6 Regional plot and correlation heatmap of CpGs located in the MAGI2 gene locus
from the regional analysis. ... 171 Figure 4. 7 Regional plot and correlation heatmap of CpGs located in the SKAP2 gene locus
from the regional analysis. ... 172 Figure 4. 8 Regional plot and correlation heatmap of CpGs located in the BMPR1B gene
locus from the regional analysis. ... 173 Figure 4. 9 Associations between placental DNAm levels between A) cg22593959 and
FAM3C mRNA levels and B) cg22436429 and TFAP2E mRNA levels. Spearman’s correlation tests were computed for each significant epimutations in the Gen3G cohort and mRNA levels of nearby or covering genes quantified relatively to the endogenous control YWHAZ. ... 175 Figure 4. 10 Association between placental DNAm levels at significant CpG sites and early-childhood adiposity in the independent 3D birth cohort. ... 178
LISTE DES TABLEAUX
Tableau 1.1 Catégorisation de la surcharge pondérale basée sur le calcul d’indice de masse
corporelle et le niveau de risques de comorbidités associé ... 4
Tableau 1.3 Liste des gènes impliqués dans l’obésité monogénique ... 8
Tableau 1.4 Liste des loci causant la forme syndromique d’obésité ... 10
Tableau 1.5 Liste des polymorphisme associés à l’obésité polygénique ... 12
Table 2.1 Clinical characteristics of the mothers and offspring from first trimester of pregnancy to 5 years of age. ... 80
Table 2.2 Comparison of placental DNA methylation levels at LPL locus between exposed and non-exposed to GDM. ... 83
Table 2.3 Comparison of children’s growth between exposed and non-exposed to GDM while fœtal development. ... 84
Table 3.1 Clinical characteristics of the mother-child dyads from the Gen3G cohort. ... 125
Table 3.2 Correlations between placental LEP DNA methylation levels and cord blood leptinemia. ... 127
Table 3.3 Associations between placental LEP DNA methylation levels and markers of childhood adiposity. ... 128
Table 3.4 Associations between placental LEP DNA methylation levels and maternal glycemia during pregnancy. ... 130
Table 4.1 Characteristics of the mothers and children from Gen3G prospective cohort included in the analysis ... 162
Table 4.2 Adjusted difference in skinfold thickness in early-childhood with a 1% change in placental DNA methylation levels ... 165
Table 4.3 Regional placental DNA methylation associated to early-childhood adiposity . 169 Table 4.4 Association between DNAm levels at both single-site and epigenomic regions, and transcriptional activity ... 176
LISTE DES ABRÉVIATIONS 5-caC 5-carboxylcytosine 5-fC 5-fluorocytosine 5-hmC 5-hydroxyméthylcytosine 5-mC 5-méthylcytosine ADIPOQ Adiponectine
ADN Acide désoxyribonucléique
Alu Arthrobacter luteus
ARN Acide ribonucléique
ARNi ARN interférant
ARNm ARN messager
ARNnc ARN non-codant
ARNpi ARN piwi
ARNt ARN de transfert
ATP Adénosine triphosphate
Avy Agouti viable yellow
BER Base excision repair
BMPR1B Bone Morphogenetic Protein Receptor Type 1B CART Cocaine and amphetamine regulated transcript Cas9 CRISPR associated protein 9
CGi CpG island
c-HDL Cholestérol-lipoprotéine de haute densité Chromosome Xi Chromosome X inactif
CpG Cytosine-phosphate-guanine
CRISPR Clustered regulargly interspaced palindromic repeats CSM Cellule stromale mésenchymateuse
DG Diabète gestational
DMR Differentially methylated region
DNMT DNA méthyltransférases
DOHaD Developmental origins of health and diseases
DT2 Diabète de type 2
FAM3 Family with sequence similarity 3
FAM3C Sous-unité C de la Family with sequence similarity 3
FMN1 Formine 1
GLUT Glucose transporter
GWAS Genome-wide association study
HDAC Histones déacétylases
ICE Imprint control element
IGF Insulin growth factors
Igfr2 Insulin-like growth factor 2 receptor IMC Indice de masse corporelle
INSPQ Institut National de Santé Publique du Québec
INS-R Insulin receptor
IRS-1 Insulin receptor substrate 1
LEPR Leptin receptor
LINE1 Long interspersed nuclear element
LPL Lipoprotéine lipase
LTR Répétitions terminales longues
mADN Méthylation de l'ADN
MAGI2 Membrane Associated Guanylate Kinase, WW And PDZ Domain Containing 2 MC4R Récepteur de la mélanocortine – 4
miARN Micro ARN
mQTL Methylation quantitative trait locus OMS Organisation mondiale de la santé
Pb Paire de bases
PC1 Prohormone convertase – 1
POMC Pro-opiomélanocortine
RISC RNA-induced silencing complex
SAM S-adénylméthionine
SKAP2 Src Kinase Associated Phosphoprotein 2
SNP Polymorphisme
SS Plis cutané supra-scapulaire
STZ Steptozotocine
TDG ADN thymine glycosylase
TET Ten eleven translocation
TFAP2E Sous-unité E de la Transcriptional regulation by the AP-2 TR Plis cutané tricipital
TSS Transcription start site
α-MSH Hormone stimulante α-mélanocyte
CHAPITRE 1 - INTRODUCTION
1.1. Le portrait de l’obésité
L’obésité est aujourd’hui considérée comme un fléau majeur en matière de santé. Dans les faits, l’obésité est en constante progression depuis la deuxième guerre mondiale avec une augmentation plus marquée depuis 1975 : depuis la prévalence a presque triplé (Caballero 2007). En 2015, près de 10% de la population mondiale était considérée obèse (Friedrich 2017). Alors que le nombre d’individus obèses semble s’être relativement stabilisé dans certains pays industrialisés, la prévalence du surpoids (embonpoints ou obésité) a poursuivi son augmentation alarmante ailleurs dans le monde (Hruby and Hu 2015). Par exemple, au Canada, la prévalence d’adultes souffrant d’obésité est passée de 14% à 24% (Statistics-Canada 2019; PHAC 2011).
Cependant, on observe une grande hétérogénéité de la prévalence, même à l’échelle provinciale. Au Québec, la prévalence du surpoids chez les adultes est passée de 35% à 51% entre 1987 et 2010 selon le rapport de l’Institut National de Santé Publique du Québec (INSPQ) (Martel 2014). Les données de recensement de 2015 démontrent que parmi les 60% de québécois adultes en surpoids, 23% souffraient d’obésité, ce qui est légèrement plus faible que dans le reste du Canada (29%) (Statistics-Canada 2019). Ces mêmes données permettent également de constater que le dimorphisme sexuel associé à la prévalence d’obésité est plus modeste au Québec en comparaison avec le reste du Canada (écart de 1% au Québec vs 4% au Canada). Au final, il est possible de dresser un portrait du surpoids et de l’obésité au Québec et au Canada. Il est toutefois important de noter qu’aucune de ces données n’a été
ajustée pour les facteurs confondants susceptibles d’expliquer la variabilité observée (e.g. origine ethnique) (Kim et al. 2018).
1.1.1. La définition de l’obésité
Les travaux des dernières décennies ont permis de mieux comprendre la physiopathologie de l’obésité. Brièvement, elle se définit par une accumulation excessive de masse grasse (ou masse adipeuse), affectant la qualité de vie de l’individu atteint. Par conséquent, le diagnostic clinique est relativement simple puisqu’il se base sur des marqueurs anthropométriques tel que l’indice de masse corporelle (IMC). L’IMC est le principal outil diagnostic de l’obésité préconisé par l’Organisation mondiale de la santé (OMS) (WHO 2000). Il permet de quantifier la surcharge pondérale qui est, par définition, l’excédent de poids relié à une accumulation de masse adipeuse à la suite d’un déséquilibre entre les gains et les dépenses en énergie. Il se calcule en divisant le poids (kg) par la taille au carré (m2). Ainsi, l’IMC permet de catégoriser les individus selon leur masse corporelle.
À titre d’exemple, un IMC entre 18,5 kg/m2 et 24,9 kg/m2 représente le poids idéal d’un
individu, alors qu’on catégorise les individus obèses lorsque leur IMC est supérieur à 30 kg/m2. Les différentes catégories d’IMC sont représentées dans le tableau 1.1.
Tableau 1.1 Catégorisation de la surcharge pondérale basée sur le calcul d’indice de masse corporelle et le niveau de risques de comorbidités associé
Catégorie IMC (kg/m²) Risque de
comorbidités
Maigreur <18,5 Faible
Normal 18,5 à 24,9 Moyen
Surpoids 25,0 à 29,9 Légèrement augmenté
Obésité
Type I (modérée) Type II (sévère) Type III (morbide)
30,0 à 34,9 35,0 à 39,9 ≥40,0 Modérément augmenté Fortement augmenté Très fortement augmenté
Tiré et modifié à partir de ('Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults: executive summary. Expert Panel on the Identification, Evaluation, and Treatment of Overweight in Adults' 1998)
Le tableau 1.1. présente également le niveau de risques de comorbidités associé aux différentes catégories d’IMC. En effet, l’obésité a depuis longtemps été associée à plusieurs complications cardiométaboliques incluant le diabète de type 2 (DT2), l’hypertension artérielle, les dyslipidémies, certaines maladies cardiaques, accidents ischémiques cérébraux et cancers (Jarolimova, Tagoni, and Stern 2013). En somme, l’obésité est associée à une augmentation de la morbidité et de la mortalité. À titre d’exemple, une étude réalisée auprès de canadiens adultes a démontré que près de 20% des cas de décès prématurés étaient attribuables à l’obésité (Janssen 2013). Il existe néanmoins un dimorphisme sexuel dans le risque de comorbidités associées à l’obésité. En effet plusieurs équipes de recherche ont
démontré que les femmes en surpoids sont plus à risque de souffrir de comorbidités en comparaison avec des hommes du même groupe de poids, mais on ignore encore les causes de cette discordance (Jorm et al. 2003; Mather et al. 2009; Calza, Decarli, and Ferraroni 2008; Imai et al. 2008).
1.1.2. L’étiologie et les facteurs de risque
D’un point de vue étiologique, il est bien connu que l’homéostasie énergétique résulte d’un équilibre entre les apports et les dépenses énergétiques. Autrefois on attribuait entièrement le développement de l’obésité à des habitudes alimentaires malsaines et excessives. Cependant, les données probantes les plus récentes suggèrent que l’étiologie est beaucoup plus complexe.
Dans les faits, l’alimentation permet de fournir l’apport énergique; l’excédent énergétique sera emmagasiné sous forme de triacylglycérols dans les vacuoles lipidiques des adipocytes (Spiegelman and Flier 2001). À l’échelle de l’histoire de l’Homme, cette capacité d’emmagasiner l’énergie permettait de traverser les longues périodes de famines (Neel 1999). En effet, les triacylglycérols permettaient de survivre en période de carence énergétique puisqu’ils sont très riches en énergie, notamment parce qu’ils sont emmagasinés sous forme d’anhydres (pauvre en eau) (Ahmadian et al. 2007). En effet, chaque gramme de graisse contient 37,6 kJ, contrairement aux carbohydrates et aux protéines qui n’en contiennent que 16,7 (Rolls 2000). De nos jours, il n’y a presque plus de périodes de famines et cet avantage évolutif est donc devenu une inadaptation du métabolisme énergétique. Le développement de l’obésité se fait généralement de manière progressive. Contrairement aux croyances, ce ne sont effectivement pas les grands écarts entre les apports et les dépenses énergétiques (qui surviennent rarement) qui sont le plus nuisibles, mais l’effet cumulatif des petits
déséquilibres de l’homéostasie énergétique. Les études longitudinales et transversales permettent de documenter ce phénomène. Hill et ses collègues ont rapporté que le gain de poids moyen est d’une à deux livres par an durant 20 ans (Hill et al. 2003). Ils ont d’ailleurs estimé que ce gain de poids est attribuable à un déséquilibre énergétique de seulement 15 kcal/jour. À titre de comparaison, une pomme de taille moyenne correspond à environ 90 kcal. Néanmoins, la variabilité de réponses à une suralimentation suggère l’implication de facteurs étiologiques intrinsèques (i.e. génétique) et extrinsèques (i.e. environnement et développement psycho-social) (Weinsier et al. 1998).
1.1.2.1. Les facteurs intrinsèques
La contribution de facteurs génétiques au développement de l’obésité et la distribution de la masse adipeuse n’est plus à démontrer. Cela fait déjà plusieurs décennies qu’on a observé des différences d’adaptations interindividuelles en réponse à une suralimentation (à court ou à long terme) (Forbes et al. 1986; Sims 1986; Sims et al. 1968; Salans, Horton, and Sims 1971; Sims et al. 1973; Sims et al. 1972). Les preuves les plus convaincantes de la contribution génétique ont été fournies par des cohortes de frères et sœurs. Ainsi, en 1986, Stunkard et ses collègues ont publié des résultats démontrant que les enfants adoptés avaient un IMC plus fortement corrélé à celui de leurs parents biologiques qu’à celui de leurs parents adoptifs (Stunkard et al. 1986). Des études réalisées dans des cohortes de jumeaux ont permis d’estimer les pourcentages de masse grasse et d’IMC attribuables aux facteurs génétiques. Alors qu’ils sont identiques d’un point de vue génétique, 70-90% de la variabilité de la masse grasse est expliquée par la génétique chez les jumeaux monozygotes, contrairement à un 35-45% pour les jumeaux dizygotes (Hebebrand et al. 2003; Farooqi and O'Rahilly 2005b; Bell, Walley, and Froguel 2005). L’héritabilité de l’IMC
pourrait même avoisiner les 77-84% (Stunkard, Foch, and Hrubec 1986). Les travaux les plus importants ont été réalisés au Québec et ont démontré que des jumeaux identiques soumis à une même diète hypercalorique avaient un gain de poids et une distribution de la masse adipeuse similaires au terme de 100 jours et 84 000 kilocalories excédentaires (Bouchard et al. 1990). Il est toutefois important de noter que le devis expérimental et les méthodes utilisées pour estimer l’héritabilité de l’obésité varient considérablement, ce qui peut expliquer la variabilité de la contribution génétique obtenue dans ces différentes études. Quoiqu’il en soit, ces résultats supportent l’héritabilité de l’obésité, bien que le pourcentage réel ne soit pas encore clairement élucidé (Stunkard, Foch, and Hrubec 1986; Maes, Neale, and Eaves 1997). L’identification de la génétique comme facteurs étiologiques de l’obésité a néanmoins permis de classifier l’obésité selon l’étiologie génétique (monogénique, syndromique et polygénique).
1.1.2.1.1. L’obésité monogénique
Des études réalisées chez des rongeurs ont permis d’identifier les différents facteurs génétiques responsables de l’obésité monogénique (Farooqi and O'Rahilly 2005a). En effet, dans de rares cas, l’obésité peut résulter de variants génétiques rares, transmis de manière autosomale récessive, qui engendrent une forme précoce et plus sévère (i.e. communément associée à des altérations du système endocrinien) (Ramachandrappa and Farooqi 2011). Ces variants rares sont généralement localisés dans des gènes qui contribuent de manière majeure au phénotype. Par exemple, la forme monogénique d’obésité identifiée à ce jour affecte principalement le système leptinergique – mélanocortinergique, qui permet de réguler la satiété et la perte de poids (Hinney, Vogel, and Hebebrand 2010). La liste des gènes impliqués aujourd’hui dans le développement de l’obésité monogénique est présentée dans
le Tableau 1.3. Il faut toutefois noter qu’il n’est pas possible d’écarter les facteurs environnementaux et les interactions gène-gène comme facteurs étiologiques de l’obésité monogénique (Lalouel et al. 1983; Hinney, Vogel, and Hebebrand 2010).
Tableau 1.2 Liste des gènes impliqués dans l’obésité monogénique
Locus Protéine encodée Fonctions physiologiques habituelles LEP Leptine Hormone sécrétée par les adipocytes qui
régule les comportements liés à l’alimentation
LEPR Récepteur de la leptine Se fixe à la leptine pour activer la synthèse de POMC
POMC Pro-opiomélanocortine Précurseur protéique de l’hormone stimulante α-mélanocyte (α-MSH) et des autres protéiques hormonales
PC1 Prohormone convertase – 1 Catalyse le clivage de POMC en α-MSH MC4R Récepteur de la mélanocortine -
4
Fixe MC4R au récepteur α-MSH, exprimé dans l’hypothalamus pour activer les signaux anorexigéniques
Tiré (et traduit) à partir de Rao, Lal, and Giridharan 2014. 1.1.2.1.2. L’obésité syndromique
La forme syndromique se manifeste également en bas âge. Elle est causée par des altérations génétiques ou chromosomiques qui affectent simultanément plusieurs gènes et qui causent des syndromes spécifiques. Ces gènes et les syndromes associés sont détaillés dans le Tableau 1.4. Contrairement à la forme d’obésité monogénique, la forme syndromique est souvent associée à des malformations congénitales et des désordres neurologiques (e.g. retard
de développement et intellectuel) (D'Angelo et al. 2013; Beales et al. 1999; Milani et al. 2014).
Tableau 1.3 Liste des loci causant la forme syndromique d’obésité
Transmission Syndrome Locus Gène
Autosomal dominant
Syndrome Prader-Willi (PWS) 15q11.2-q12 - Osteodystrophie héréditaire de Albright 20q13.2 GNAS1
Syndrome du X fragile Xq27.3 FMR1 Syndrome ulnaire-mammaire 12q24.1 TBX3 Autosomal récessif Syndrome Bardet-Biedl 11q13 16q21 15q22 20p12 BBS1 BBS2 BBS4 BBS6
Syndrome Alström 2p13 ALMS1
Syndrome Cohen 8q22 - Lié à l’X Syndrome Börjeson-Forssman-Lehmann Xq26 PHF6 Syndrome MEHMO Xq22.13 - Simpson-Golabi-Behmel, type 2 Xp22 - Syndrome Wilson-Turner Xp21.2 -
Tiré (et traduit) à partir de Rao, Lal, and Giridharan 2014.
1.1.2.1.3. L’obésité polygénique ou communce
La forme polygénique est la plus fréquente forme d’obésité. Les premières études visant à identifier les facteurs génétiques impliqués dans l’étiologie de cette forme d’obésité reposent sur des études de liaison (linkage analysis) et des études d’association par gène candidat. Bien que ces méthodes aient permis d’identifier les principaux facteurs génétiques impliqués dans les formes monogéniques et syndromiques, elles n’ont pas donné les résultats escomptés pour la forme polygénique pour diverses raisons (e.g. faible reproductibilité,
manque de puissance statistique, hétérogénéité des populations, coût et temps du génotypage) (Rankinen et al. 2006; Saunders et al. 2007; Herrera and Lindgren 2010).
Avec les avancées technologiques, les études d’associations pangénomiques (genome-wide association studies; GWAS) ont permis d’identifier près de 100 loci associés à l’IMC dans l’enfance (5-6 ans) (Herrera and Lindgren 2010; Couto Alves et al. 2019) (voir Tableau 1.5). Ces techniques se basent sur le déséquilibre de liaison des polymorphismes (SNP) pour limiter le nombre de variants génétiques testés. De ce fait, identifier le variant causal associé au phénotype demeure un véritable défi ('A haplotype map of the human genome' 2005). Les efforts pour identifier les déterminants génétiques de l’obésité se poursuivent, il se peut que la liste présentée en Tableau 1.5 ne soit plus à jour. Quoiqu’il en soit, ces variants génétiques n’expliquent pas plus de 2% de la variabilité interindividuelle de l’IMC (Lindgren et al. 2009; Rao, Lal, and Giridharan 2014). Il reste donc encore beaucoup à faire afin de parvenir à identifier tous les facteurs qui expliquent pourtant 40-70% d’héritabilité de l’obésité (i.e. missing heritability).
Tableau 1.4 Liste des polymorphismes associés à l’obésité polygénique
Polymorphisme
(SNP) Chromosome Gène le plus proche
rs1000940 17 RABEP1 rs10132280 14 STXBP6 rs1016287 2 LINC01122 rs10182181 2 NCOA1 rs10733682 9 LMX1B rs10938397 4 GNPDA2 rs10968576 9 LINGO2 rs11030104 11 BDNF rs11057405 12 CLIP1 rs11126666 2 KCNK3 rs11165643 1 PTBP2 rs11191560 10 NT5C2 rs11583200 1 ELAVL4 rs1167827 7 HIP1 rs11688816 2 EHBP1 rs11727676 4 HHIP rs11847697 14 PRKD1 rs12016871 13 MTIF3 rs12286929 11 CADM1 rs12401738 1 FUBP1 rs12429545 13 OLFM4 rs12446632 16 GPRC5B rs12566985 1 FPGT-TNNI3K rs12885454 14 PRKD1 rs12940622 17 RPTOR rs13021737 2 TMEM18 rs13078960 3 CADM2 rs13107325 4 SLC39A8 rs13191362 6 PARK2 rs13201877 6 IFNGR1 rs1441264 13 MIR548A2 rs1460676 2 FIGN rs1516725 3 ETV5 rs1528435 2 UBE2E3 rs1558902 16 FTO rs16851483 3 RASA2 rs16907751 8 ZBTB10
rs16951275 15 MAP2K5 rs17001654 4 SCARB2 rs17024393 1 GNAT2 rs17094222 10 HIF1AN rs17203016 2 CREB1 rs17405819 8 HNF4G rs17724992 19 PGPEP1 rs1808579 18 C18orf8 rs1928295 9 TLR4 rs2033529 6 TDRG1 rs2033732 8 RALYL rs205262 6 C6orf106 rs2075650 19 TOMM40 rs2080454 16 CBLN1 rs2112347 5 HMGCR/COL4A3BP rs2121279 2 LRP1B rs2176040 2 LOC646736 rs2176598 11 HSD17B12 rs2207139 6 TFAP2B rs2245368 7 PMS2L11 rs2365389 3 FHIT rs2650492 16 SBK1 rs2820292 1 NAV1 rs2836754 21 ETS2 rs29941 19 KCTD15 rs3101336 1 NEGR1 rs3736485 15 DMXL2 rs3810291 19 ZC3H4 rs3817334 11 MTCH2/C1QTNF4/SPI1 rs3849570 3 GBE1 rs3888190 16 ATXN2L/SBK1/SULT1A2/TUFM rs4256980 11 TRIM66 rs4740619 9 C9orf93 rs4787491 16 INO80E rs492400 2 USP37 rs543874 1 SEC16B rs6091540 20 ZFP64 rs6465468 7 ASB4 rs6477694 9 EPB41L4B rs6567160 18 MC4R rs657452 1 AGBL4
rs6804842 3 RARB rs7138803 12 BCDIN3D rs7141420 14 NRXN3 rs7164727 15 LOC100287559 rs7239883 18 LOC284260 rs7243357 18 GRP rs758747 16 NLRC3 rs7599312 2 ERBB4 rs7715256 5 GALNT10 rs7899106 10 GRID1 rs7903146 10 TCF7L2 rs9374842 6 LOC285762 rs9400239 6 FOXO3 rs9540493 13 MIR548X2 rs9641123 7 CALCR rs977747 1 TAL1 rs9914578 17 SMG6 rs9925964 16 KAT8 rs2287019 19 QPCTL
Tiré de Couto Alves et al. 2019
1.1.2.2. Les facteurs extrinsèques
Il apparaît évident que certaines expositions environnementales pourraient influencer la pathogenèse de l’obésité. Par exemple, les individus ayant une prédisposition génétique pourraient avoir un plus grand risque de développer de l’obésité lorsqu’exposés à un environnement obésogène (favorisant un débalancement énergétique) (Ogden et al. 2007). Cependant et la génétique est relativement stable au fil du temps (et même de manière intergénérationnelle). L’accroissement de la prévalence de l’obésité durant les dernières décennies est donc potentiellement davantage attribuable à des changements environnementaux variés (i.e. habitudes de vie).
D’abord, l’alimentation a considérablement changé au fil du temps, plus particulièrement dans les pays industrialisés (Jeffery and Harnack 2007). Brièvement, les
principaux changements susceptibles d’augmenter les risques d’obésité concernent la teneur inadéquate en macronutriments (glucides, lipides et protéines), la grande densité énergétique des aliments, la taille démesurée des portions et la présence de sucres ajoutés dans les breuvages (Yancy, Wang, and Maciejewski 2014; Ello-Martin, Ledikwe, and Rolls 2005; Benton 2015; Malik, Schulze, and Hu 2006).
Par ailleurs, la diminution des tâches nécessitant une dépense énergétique et les changements technologiques (e.g. ordinateur, télévision, jeux vidéo) ont également contribué à augmenter la prévalence de l’obésité. La sédentarité est un facteur obésogène notamment parce qu’elle est associée à l’augmentation de l’IMC ou à une diminution de la dépense énergétique et au développement de l’obésité dans plusieurs études (Malik, Schulze, and Hu 2006; Gupta et al. 2016; Vasques et al. 2012; Siddarth 2013). L’alimentation et la sédentarité contribuent à la pathogenèse de l’obésité, et ce pour toutes les catégories d’âges.
La naissance et les premiers mois de vie peuvent également avoir des répercussions considérables sur la santé des individus à long terme. Ainsi, l’association entre l’accouchement par césarienne et le risque d’obésité a été rapportée dans plusieurs études (Kuhle, Tong, and Woolcott 2015; Magne et al. 2017; Barros et al. 2017; Ahlqvist et al. 2019; Yuan et al. 2016; Chavarro et al. 2020). En effet, l’acquisition du microbiome intestinal (i.e. ensemble de bactéries composant le système digestif) pourrait être altérée par le manque d’exposition au microbiome vaginal maternel (Kuhle, Tong, and Woolcott 2015). Puisque le microbiome intestinal contribue à une multitude de processus essentiels (e.g. inflammation, absorption de nutriments, développement du système immunitaire) impliqués dans l’équilibre énergétique, l’accouchement par césarienne pourrait donc augmenter le risque d’obésité (Kuhle, Tong, and Woolcott 2015).
Des études ont également mis en évidence l’importance de l’alimentation néonatale. Bien que la contribution du microbiome intestinal soit à nouveau susceptible d’expliquer une partie de cette relation, l’association entre l’alimentation par une méthode autre que l’allaitement et l’obésité est moins convaincante (Rossiter et al. 2015; Oddy et al. 2014; Kramer et al. 2009; Vehapoglu et al. 2014; Fergusson, McLeod, and Horwood 2014). La divergence des résultats pourrait résulter, entre autres, de l’hétérogénéité des approches analytiques (i.e. covariables incluses dans les modèles). À titre d’exemple, dans les travaux de Fergusson et ses collègues, des analyses de médiations ont permis de démontrer que la relation entre l’alimentation néonatale et l’IMC à l’âge adulte (i.e. 30-35 ans) était médiée par la croissance néonatale (Fergusson, McLeod, and Horwood 2014). Or, cette variable (en plus de toutes celles qui auraient pu être considérées) n’a pas été incluse dans les modèles statistiques des autres études citées précédemment.
1.1.2.2.1. L’obésité infantile
L’obésité infantile est aujourd’hui reconnue comme l’un des facteurs de risque de l’obésité à l’âge adulte. En effet, l’obésité infantile et l’obésité à l’âge adulte ne sont pas complètement indépendants puisque près de 80% des enfants obèses le demeureront à l’âge adulte (Freedman et al. 2005; Singh et al. 2008; Lee et al. 2010). Ce phénomène pourrait s’expliquer par le fait que l’expansion du tissu adipeux exacerbée dans l’enfance par l’hyperplasie et l’hypertrophie des adipocytes, tend à persister à l’âge adulte, bien que cette théorie spéculative n’aie pas été testée suffisamment (Freedman and Sherry 2009). Des études ont également rapporté qu’un gain de poids rapide en bas âge pouvait être un prédicteur d’obésité à l’âge adulte (Bjerregaard et al. 2014; Rolland-Cachera et al. 1984; Ekelund et al. 2006; Stettler et al. 2003; Baird et al. 2005). Cette association entre la prise de
poids et l’obésité n’a toutefois été démontrée que pour les enfants en bas âge. Des fluctuations de poids dans l’enfance et à l’âge adulte semblent également augmenter le risque d’obésité et ses comorbidités, bien que cette relation puisse être réversible par une perte de poids, même non-constante (Dyer, Stamler, and Greenland 2000; Folsom et al. 1996; French et al. 1997; Lissner et al. 1991; Stevens and Lissner 1990; Rzehak et al. 2007; Thorpe et al. 2013; Knowler et al. 2009; Li et al. 2015).
À titre informatif, l’IMC chez l’enfant est rapporté à une courbe de croissance séparée par sexe et par tranches d’âge (score z d’IMC). Bien qu’il en existe plusieurs, la plus utilisée est la charte de croissance de l’OMS (de Onis et al. 2012). Cette courbe permet d’évaluer la surcharge pondérale de l’enfant en se basant sur le percentile d’IMC, mais il n’y a pas de consensus international sur la valeur diagnostic seuil (>95 ou >97e percentile) (Freedman and
Sherry 2009). À l’IMC peuvent s’ajouter des mesures de composition corporelle comme la circonférence de la taille, l’épaisseur des plis cutanés, l’absorptiomètrie à rayons X à double énergie (DEXA) et l’impédance bioélectrique, mais ces outils sont plutôt utilisés dans un but de phénotypage ou de suivi des études et non pas de diagnostic, (Wells and Fewtrell 2006).
Il apparaît toutefois essentiel de dire que l’obésité infantile est une maladie à part entière et que ce désordre métabolique est encore plus préoccupant chez les enfants (en comparaison des adultes) en considérant ses répercussions à court ou à long terme : les complications sont d’ordre psycho-social (e.g. marginalisation sociale et isolement, faible estime), pulmonaires (e.g. asthme, apnée du sommeil), cardiovasculaires (e.g. hypertension, athérosclérose), métaboliques (e.g. résistance à l’insuline, DT2, dyslipidémies), gastrointestinales (e.g. reflux gastrique, stéatose hépatique non-alcoolique),
musculosquelettiques (e.g. Tibia vara ou maladie de Blount, épiphyse fémorale-glissante) (Reilly et al. 2003; Daniels 2009; Sahoo et al. 2015).
1.2. Le portrait de l’obésité infantile
L’obésité infantile est sujet de préoccupation à l’échelle mondiale. En effet, pas moins de 41 millions d’enfants de moins de 5 ans souffraient de surpoids (embonpoint et obésité) en 2016 et les modèles prédictifs estiment que plus de 70 millions d’enfants souffriront de ce désordre métabolique d’ici 2025. (WHO 2017). Bien que l’obésité ait longtemps été considérée comme un problème relié aux pays industrialisés, les chiffres actuels démontrent clairement que les pays en voie de développement n’y échappent pas (Ng et al. 2014). En Amérique du nord, le tiers des enfants souffre d’embonpoint ou d’obésité, et les proportions au Canada avoisinent un sur quatre (9% diagnostiqués obèses) (Rao et al. 2016). La situation au Québec est très similaire à celle du Canada avec environ 25% des enfants (26% chez les garçons et 24% chez les filles) en surpoids. Le dimorphisme sexuel est toutefois très frappant pour la prévalence québécoise de l’obésité infantile avec un 13% de garçons atteints en comparaison du 6% de prévalence chez les filles (INSPQ 2016).
1.2.1. Le rapport de commission pour mettre fin à l’obésité infantile
En 2016, la collaboration de plusieurs nations a permis d’émettre un rapport de commission sur les moyens de mettre un terme à l’épidémie croissante d’obésité infantile, qui a été approuvé par l’OMS (WHO 2017). Ce rapport présente plusieurs recommandations en matière de politiques gouvernementales qui visent à prévenir l’obésité infantile, de la naissance à l’adolescence. Les recommandations peuvent se résumer à 1) surveiller, 2) traiter et 3) prévenir l’obésité infantile.
1) Surveiller; il apparaît essentiel de miser sur une réduction de l’exposition à un environnement obésogène. Pour cela, le rapport de la commission propose la mise en place de programmes favorisant une alimentation riche en aliments sains (e.g. fibres, protéines) et pauvres en aliments hypercaloriques (e.g. glucides, gras trans) ainsi que l’élaboration de programmes favorisant l’activité physique. 2) Traiter; il apparaît prioritaire, non seulement d’identifier les enfants à risque, mais surtout de traiter les enfants atteints d’obésité en vue éventuellement de briser le cycle de transmission de l’obésité d’une génération à l’autre. Bien que les traitements actuellement offerts pour contrer l’obésité infantile sont peu nombreux et se sont montrés peu efficaces à long terme (Bohler et al. 2013), il est impératif de miser sur l’éducation familiale (mode de vie et les habitudes alimentaires), pour que les familles impliquées soient en mesure de gérer leur poids. 3) Prévenir; la dernière catégorie de recommandations concerne la réduction des facteurs de risque d’obésité infantile en s’attardant principalement aux phases développementales les plus susceptibles de moduler la santé des individus donc de la période prénatale à l’adolescence.
Bien que la majorité des solutions proposées aient pour objectif de mieux guider les familles en matière de bonnes habitudes de vie, de la petite enfance à l’âge adulte, le rapport souligne également l’importance de la période pré-conceptionnelle et de la grossesse dans l’étiologie de l’obésité infantile. Il est aujourd’hui bien établi que la santé des parents a une influence sur la santé et le développement de leurs enfants. En effet, la dénutrition maternelle, l’obésité et/ou la prise de poids durant la grossesse, l’hyperglycémie maternelle, le tabagisme ou l’exposition à certaines toxines (i.e. produits chimiques xénobiotiques) sont actuellement suspectés d’être des facteurs étiologiques de l’obésité infantile (Yu et al. 2013; Eriksson et al. 2014; Okubo et al. 2014; Poston 2012; Oken, Levitan, and Gillman 2008; Janesick and
Blumberg 2011). Bien que les évidences concernant la santé des pères soient moins nombreuses, il est également important de poursuivre les travaux sur ce possible facteur de risque (McPherson et al. 2014). Dans tous ces cas, les mécanismes sous-jacents responsables de la transmission de la santé ne sont pas encore clairement élucidés.
1.1. Le développement prénatal
Il est essentiel de bien définir les étapes du développement prénatal et leur implication dans la croissance prénatale afin de bien situer cette thèse. Les trois phases distinctes du développement prénatal comprennent : 1) la phase pré-implantatoire, caractérisée par une série de mitoses et de maturations cellulaires qui s’échelonne sur les deux premières semaines de vie; 2) la phase post-implantatoire où se produit l’organogenèse jusqu’à la fin de la 8e semaine après la fertilisation (i.e. 11e semaine de gestation); et 3) la
période fœtale qui comprend la maturation des tissus et des organes et la croissance rapide du fœtus (dépôt de graisse sous-cutanée) (Figure 1.1) (Larsen 2017).
Figure 1. 1 Chronologie du développement prénatal en cours de grossesse.
Le développement prénatal se caractérise par des phases pré-implantatoire, post-implantatoire et fœtale qui comprennent la fertilisation, l’implantation, la placentation, l’organogenèse, l’adipogenèse et la croissance et maturation des tissus et organes. Ces processus s’échelonnent sur 40 semaines en moyenne. Tiré et modifié à partir de Medical gallery of Mikael Häggström 2014.
1.1.1. La phase pré-implantatoire
La période pré-implantatoire débute avec la fertilisation et permet de passer de zygote (1 cellule), au stade blastocyste (200-300 cellules). Au terme de cette phase de développement qui dure au maximum 2 semaines, le blastocyste est une masse de cellules endodermiques pluripotentes (futur embryon) entourée d’une enveloppe de cellules externes appelées trophoblastes (futur placenta) (Niakan et al. 2012).
1.1.2. La phase post-implantatoire
Suit la période post-implantatoire; elle est caractérisée par l’implantation et la placentation. En effet, le blastocyste migre d’abord de la trompe à la cavité utérine, puis s’implante dans l’endomètre lorsque ce dernier est en phase réceptive (Chavatte-Palmer and Guillomot 2007). Pour cela, le trophoblaste prolifère et se fixe à l’épithélium de l’endomètre par invasion pour former une couche épaisse de cytoplasme contenant plusieurs noyaux (syncytiotrophoblaste). Le blastocyste implanté, surplanté de sa barrière syncytiotrophoblastique devient totalement indépendant de l’utérus, du moins jusqu’à l’invasion des artères spiralées maternelles qui assurent un apport sanguin constant à l’embryon (Knofler and Pollheimer 2013). Du tissu conjonctif tapisse l’intérieur du blastocyste, résultant en une masse cellulaire interne au trophoblaste dont l’ensemble deviendra le chorion. À mesure qu’il se développe, le tissu conjonctif provenant de l’embryon forme de nombreux vaisseaux sanguins ramifiés pour donner ultérieurement des villosités choriales (Carter, Enders, and Pijnenborg 2015). La partie la plus profonde du chorion deviendra la plaque chorionique placentaire. En effet, de cette plaque emmergent de nombreuses villosités choriales dans lesquelles se sont taraudées des artères utérines (espace
intervilleux). Ainsi, au terme de la quatrième semaine de développement prénatal, la communication materno-fœtale est complètement établie. Au final, le sang fœtal traverse les vaisseaux du cordon ombilical jusqu’à la plaque chorionique, alors que la circulation maternelle, complètement indépendante de la circulation fœtale, prend place dans l’espace intervilleux (Demir et al. 1989). La communication entre les deux systèmes vasculaires se fait exclusivement par diffusion.
Parallèlement à la placentation enclenchée par les trophoblastes, les cellules endodermiques se subdivisent en épiblaste externe et en hypoblaste interne pour former le disque embryonnaire didermique (Larsen 2017). Bien que l’hypoblaste contribue au développement du mésoderme extra-embryonnaire et du sac vitellin (réserve nutritive), il n’est pas directement associé au développement embryonnaire. La transition du disque didermique en tridermique (i.e. gastrulation) débute par l’invagination de l’épiblaste vers l’intérieur de l’embryon (ligne primitive). La couche la plus profonde (la plus proche de l’hypoblaste) est l’endoderme. La couche intermédiaire est le mésoderme, alors que la couche superficielle de cellules épiblastiques qui borde la cavité amniotique constitue l’ectoderme (Larsen 2017). Ces feuillets embryonnaires sont à l’origine de tous les tissus et organes du corps humain (figure 1.2). L’organogenèse débute lorsque l’ectoderme donne naissance au tube neural, aux alentours du 20e jour post-implantation (Fang et al. 2010). Cette période,
qui s’étend sur 36 jours, est accompagné d’une perte de pluripotence des cellules qui s’engagent dans des voies de différentiations cellulaires spécifiques (i.e. « lineage comittment »). L’amalgame de spécialisations et de différenciations cellulaires résultent en la formation des organes primitifs (Yamanaka 2009). Au terme de l’organogenèse, l’embryon est appelé fœtus.
Figure 1. 2 Origine embryonnaire des tissus et organes.
L’épiblaste donne naissance aux trois feuillets embryonnaires à l’origine de tous les organes et systèmes du corps humain. 1) L’ectoderme permet de former le système oculaire (yeux), tégumentaire (peau) et nerveux. 2) Le mésoderme deviendra le système rénal, cardiaque et musculosquelettique comprenant entre autres les adipocytes. 3) L’endoderme permettra la formation des autres systèmes et organe (e.g. système pancréatique, pulmonaire, hépatique). Modifiée à partir de http://www.cancerlink.ru/enstemcells.html.
1.1.3. Le développement fœtal
La croissance fœtale comprend la maturation (à terme ou non) des organes, la programmation de certains tissus (e.g. distribution des adipocytes) et la régulation de certaines fonctions biologiques (e.g. métabolisme) (Hanson et al. 2015). Par conséquent, le bon fonctionnement de tous ces processus clés du développement est crucial non-seulement pour éviter un avortement spontané, mais également pour optimiser la santé de l’enfant à plus long terme. Le développement fœtal est orchestré par de nombreux facteurs intrinsèques (e.g. génétique, hormones fœtales) et extrinsèques (e.g. efficacité métabolique des échanges materno-placentaires, expositions maternelles et environnementales) (Hanson et al. 2015).
Ainsi, utiliser une approche holistique des déterminants de la croissance fœtale est désormais essentiel : il est primordial d’investiguer les potentielles interactions entre les facteurs génétiques et environnementaux qui peuvent affecter le développement et la croissance fœtale (Gluckman and Hanson 2004).
1.2. Les origines développementales de la santé
Des preuves solides démontrent que l’exposition à certains facteurs environnementaux durant le développement prénatal augmente la susceptibilité de développer des maladies chroniques à long terme (Barker and Osmond 1986). En effet, dès les années 1970, des travaux avaient démontré que le risque d’obésité à l’âge adulte peut être modulé par une période de famine sévère durant la grossesse (Ravelli, Stein, and Susser 1976). En effet, Ravelli et ses collègues avaient étudié plus de 300 000 hommes d’âges adultes nés pendant la période de famine sévère qui a fait rage aux Pays-Bas durant l’hiver 1944-45. Ils avaient observé que les individus exposés à la famine in utero durant le premier trimestre avaient une susceptibilité accrue à développer de l’obésité, alors que si l’exposition survenait au cours du dernier trimestre de grossesse, le risque était réduit.
Les travaux subséquents de Barker et Osmond ont démontré que les conséquences d’une exposition précoce à la malnutrition ne se limitait pas à une augmentation du risque d’obésité, mais aussi à une augmentation de la susceptibilité à développer des maladies cardiovasculaires (e.g. ischémie myocardique) ou métaboliques (e.g. obésité, syndrome métabolique, diabète) (Barker and Osmond 1986). Ils ont également souligné la corrélation négative entre le poids à la naissance et le risque de mortalité associée à une ischémie myocardique (Barker and Osmond 1986; Barker et al. 1989). C’est sur la base de ces résultats qu’ils ont bâti l’hypothèse du phénotype économe (Thrifty phenotype). En effet, cette
dernière stipule qu’en réponse à un environnement intra-utérin sous-optimal, le fœtus se « programme » par le biais de mécanismes adaptatifs en prévision d’un environnement post-natal similaire (Barker et al. 1993; Hales and Barker 1992). Ils ont été les premiers à rapporter qu’en réponse à un apport sous-optimal en nutriments, le fœtus adapte son métabolisme et sa physiologie, ce qui peut entraîner des défauts de structure et de fonctionnement plus tard dans la vie. Ces résultats suggèrent que l’organisme a une certaine mémoire de ce qui est survenu durant le développement prénatal.
C’est sur cette prémisse qu’est né le concept de l’origine développementale de la santé et des maladies (Developmental Origins of Health and Diseases, DOHaD) qui stipule que la santé est modulée, du moins partiellement, par certains facteurs environnementaux durant des périodes critiques du développement (i.e. développement embryonnaire et fœtal) (Haugen et al. 2015). Ce concept, qui met en évidence la plasticité de l’organisme durant le développement fœtal et les répercussions post-natales potentielles des altérations fonctionnelles, est aussi connu comme la programmation fœtale. Néanmoins, il est important de considérer qu’à la base, la programmation fœtale n’est pas un mécanisme adaptatif physiopathologique (Gluckman, Hanson, and Beedle 2007). Les adaptations durant le développement fœtal normal visent plutôt à optimiser la santé de l’individu en se basant sur un environnement intra-utérin normal (Gluckman, Hanson, and Beedle 2007; Gluckman and Hanson 2004). Or, la programmation fœtale peut devenir dommageable lorsqu’il y a discordance entre l’environnement intra-utérin et post-natal (e.g. famine/abondance, abondance/famine)(Gluckman and Hanson 2004; Gluckman, Hanson, and Beedle 2007; Bateson et al. 2004; Gluckman et al. 2005; Gluckman and Hanson 2007).
À ce jour, le concept de programmation fœtale a été corroboré par quelques études dont la majorité ont été réalisées avec des modèles animaux (Clausen et al. 2009). Il est donc primordial de diriger les recherches vers des cohortes de participants humains afin de parvenir à identifier les mécanismes biologiques impliqués dans la programmation fœtale de l’obésité infantile. Le modèle d’hyperglycémie maternelle et de diabète gestationnel (DG) semblent être parmi les plus prometteurs.
1.2.1. Le portrait et la prévalence du diabète gestationnel
Le DG est l’une des plus fréquentes complications de grossesse avec une prévalence mondiale se situant entre 2-20% (Galtier 2010). Au Québec, la prévalence du DG a presque triplé entre 1989 et 2012 en passant de 2,5% à près de 8% (Chun 2017). De nombreux facteurs peuvent expliquer ces écarts de prévalence: hétérogénéité et sévérité des critères diagnostics, ethnicité, âge maternel, histoire familiale de diabète et poids à la naissance de la mère (Galtier 2010; Ben-Haroush, Yogev, and Hod 2004). Le surpoids et l’obésité maternelle sont également des facteurs de risque de DG, et se classent même en tête de liste, mais ensemble, ils n’ont été associés qu’à 20% des cas de DG (Kim et al. 2010; Kim et al. 2013).
1.2.1.1. L’homéostasie glucidique durant la grossesse
La grossesse met en branle de nombreux changements réversibles d’ordre physiologique, métabolique, biochimique, hématologique et immunologique, en vue d’assurer la croissance fœtale (Sonagra et al. 2014). Entre autres, la deuxième moitié de la grossesse s’accompagne d’une insulino-résistance périphérique, touchant principalement le tissu adipeux et les muscles squelettiques (Hunter and Garvey 1998). Ainsi, au début du troisième trimestre, la sensibilité à l’insuline est diminuée d’environ 50% (McLachlan et al. 2006). Ce mécanisme physiologique vise à graduellement économiser les carbohydrates au
profit du fœtus (en phase de croissance), grâce à une priorisation de l’utilisation maternelle des graisses plutôt que du glucose (Sivan et al. 1999).
Bien que le mécanisme d’activation ne soit pas encore élucidé, de nombreux facteurs contribuent à ce phénomène naturel; entre autres, 1) l’inactivation de la voie signalétique de l’insuline via une diminution de la phosphorylation du récepteur à insuline (INS-R) et de l’expression génique de son substrat (insulin receptor substrate 1, IRS-1) et 2) l’inhibition du transport du glucose (glucose transporter, GLUT, sous-exprimé), possiblement en réponse à de fortes variations d’hormones placentaires et maternelles (Barbour et al. 2007; Wada et al. 2010). L’insulino-résistance durant la grossesse s’accompagne d’une augmentation de la sécrétion d’insuline en vue de garder intacte l’homéostasie glucidique (i.e. euglycémie) (Catalano, Drago, and Amini 1998). Cette hyperinsulinémie, prédominante à l’état post-prandial, pourrait être engendrée par l’insulino-résistance hépatique, bien que cette hypothèse n’ait pas encore été validée. Quoiqu’il en soit, les niveaux d’insuline plasmatiques atteignent leur maximum lors de la 36e semaine de grossesse alors qu’ils sont près de 2,5 fois plus élevés
que la normale (Marcinkevage and Narayan 2011). Dans certains cas, la production d’insuline n’est pas suffisante pour contrebalancer l’insulino-résistance et ne permet pas de conserver l’homéostasie glucidique (Figure 1.3) (Marcinkevage and Narayan 2011). Ce dérèglement du glucose sanguin est à l’origine du continuum hyperglycémie-DG.
Figure 1. 3 Variations des niveaux d'insuline en cours de grossesse normoglycémique et avec diabète gestationnel.
La grossesse s’accompagne d’une insulino-résistance naturelle et réversible où les niveaux d’insuline sécrétés permettent de maintenir l’état euglycémique. Dans le cas des grossesses compliquées par un diabète gestationnel, les niveaux d’insuline entre la 24e semaine de
grossesse et l’accouchement ne permettent pas de ramener l’équilibre. Traduit de Marcinkevage and Narayan 2011.
1.2.1.2. L’insulino-résistance dans le diabète gestationnel
Les mécanismes physiopathologiques qui expliquent pourquoi seulement une partie des grossesses se détériore en DG sont encore essentiellement incompris. Deux mécanismes ont été suggérés pour expliquer la résistance à l’insuline anormale qui caractérise certaines grossesses: 1- une inefficacité de la liaison de l’insuline à INS-R et 2- des anomalies dans la voie signalétique de l’insuline.
La première hypothèse qui suggérait que l’insulino-résistance pourrait s’expliquer par une diminution de l’affinité de l’insuline pour son récepteur a été investiguée, entre autres dans les adipocytes et dans le sang. Or, les résultats non consensuels dans les adipocytes
(Beck-Nielsen et al. 1979; Andersen and Kuhl 1986, 1987, 1988; Hjollund et al. 1986) et la démonstration de Ryan et ses collègues que la fixation de l’insuline aux érythrocytes est similaire chez les femmes enceintes (avec et sans DG) ou non-enceintes (Ryan, O'Sullivan, and Skyler 1985) ont rapidement permis d’éliminer cette hypothèse.
La communauté scientifique s’est donc penchée sur les autres éléments de la cascade de signalisation de l’insuline. Brièvement, au niveau du muscle, le DG pourrait résulter d’une diminution de l’activité de la tyrosine kinase (phosphorylation d’INS-R), une surexpression de la glycoprotéine 1 (PC1) (inhibe l’action de la tyrosine kinase), une diminution de l’expression d’IRS-1 et des transporteurs de glucose 1 et 4 (GLUT1, GLUT4) (Friedman et al. 1999; Sbraccia et al. 1991; Shao et al. 2000; Yamashita, Shao, and Friedman 2000; Colomiere, Permezel, and Lappas 2010). Ensemble, ces altérations sont à l’origine de la diminution du transport du glucose et de la synthèse du glycogène observée dans les muscles squelettiques de patients avec insulino-résistance. Dans le tissu adipeux, l’hypothèse de l’altération de la chaîne signalétique (diminution de IRS-1 et GLUT-4) de l’insuline est également documentée (Okuno et al. 1995; Garvey et al. 1998; Catalano et al. 2002).
Or, la résistance à l’insuline dans le tissu adipeux semble également être associée au métabolisme des acides gras. Vers la fin de la grossesse, les niveaux d’acides gras libres augmentent en période post-prandiale ce qui stimule la libération de l’insuline, et par le fait même induit une augmentation de 40 à 60% (vs la période pré-conceptionnelle) de l’élimination du glucose (Catalano et al. 1999). L’insuline en cours de grossesse est également moins efficace pour réduire la lipolyse, ce phénomène est accentué chez les mères avec DG (Homko et al. 1999; Catalano et al. 2002). Ainsi, les grossesses avec DG sont caractérisées par une augmentation des acides gras libres post-prandiaux. Ce
dysfonctionnement dans le métabolisme des lipides est observable même avant la grossesse comme il a été démontré dans une étude longitudinale qui visait la caractérisation du profil sanguin d’acides gras libres avant et pendant les grossesses compliquées ou non par du DG. Les niveaux basaux d’acides gras libres étaient plus élevés chez les mères avec DG, et ce, même en période pré-conceptionnelle (Catalano et al. 2002). En conséquence, l’accumulation d’acides gras pourrait être à l’origine de la lipotoxicité et la libération de marqueurs inflammatoires qui accentuent le mécanisme de résistance à l’insuline dans le tissu adipeux en cas de DG (Lumeng and Saltiel 2011; Pickup 2004; Shoelson, Lee, and Goldfine 2006). Au final, cette résistance à l’insuline (normale et exacerbée dans le cas de DG) dans les muscles et le tissu adipeux engendre inévitablement une augmentation de la néoglucogénèse hépatique pour maintenir l’euglycémie (Jin et al. 2015; Butte 2000). Ces mécanismes physiopathologiques sont résumés dans la Figure 1.4.
Figure 1. 4 Mécanismes physiologiques de régulation du glucose lors de grossesses avec ou sans diabète gestationnel.
La grossesse se caractérise par une diminution réversible de la sécrétion de l’insuline par les cellules β pancréatiques et une diminution de la sensibilité à l’insuline dans les tissus périphériques, en vue de limiter l’utilisation maternelles des carbohydrates pour permettre une bonne croissance fœtale. Dans le cas de grossesses compliquées par un DG, l’insulino-résistance dans les muscles et les adipocytes est trop importante pour les besoins actuels et entraîne une augmentation de la néoglucogenèse dans le foie. Ensemble, ces mécanismes contribuent à l’hyperglycémie maternelle qui caractérise les grossesses avec DG.
1.2.1.3. Les conséquences maternelles du diabète gestationnel
Bien que cette thèse ne porte pas sur l’influence du DG sur la santé maternelle, il importe de mentionner que les mères ayant souffert de DG durant la grossesse sont plus à