Pour l'obtention du grade de
DOCTEUR DE L'UNIVERSITÉ DE POITIERS UFR des sciences fondamentales et appliquées
Institut de chimie des milieux et matériaux de Poitiers - IC2MP (Diplôme National - Arrêté du 25 mai 2016)
École doctorale : Sciences pour l'environnement - Gay Lussac (La Rochelle) Secteur de recherche : Terre solide et enveloppes superficielles
Présentée par : Fabien Baron
Le fer dans les smectites : une approche par synthèses minérales
Directeur(s) de Thèse : Sabine Petit
Soutenue le 06 septembre 2016 devant le jury
Jury :
Président Emmanuel Tertre Professeur des Universités, Université de Poitiers Rapporteur Nicolas Mangold Directeur de recherche CNRS, Université de Nantes Rapporteur Antoine Thill Docteur, CEA, Saclay
Membre Sabine Petit Directrice de recherche CNRS, Université de Poitiers Membre Sabine Valange Maître de conférences, Université de Poitiers
Membre Olivier Grauby Maître de conférences, Université d'Aix-Marseille
Pour citer cette thèse :
Fabien Baron. Le fer dans les smectites : une approche par synthèses minérales [En ligne]. Thèse Terre solide et enveloppes superficielles. Poitiers : Université de Poitiers, 2016. Disponible sur Internet
THÈSE
Pour l'obtention du grade de
DOCTEUR DE L'UNIVERSITÉ DE POITIERS UFR des sciences fondamentales et appliquées
Institut de chimie des milieux et matériaux de Poitiers - IC2MP (Diplôme National - Arrêté du 7 août 2006)
École doctorale : Sciences pour l'environnement - Gay Lussac Secteur de recherche : Terre solide et enveloppes superficielles
Présentée par :
Fabien Baron
Le fer dans les smectites : une approche par synthèses minérales
Directrice de Thèse : Sabine Petit Soutenue le 6 septembre 2016 devant le jury
Membre du jury :
Rapporteur Nicolas Mangold Directeur de recherche CNRS, Université de Nantes
Rapporteur Antoine Thill Chercheur Dr-HDR, CEA Saclay
Examinateur Olivier Grauby Maître de conférences, Université d’Aix-Marseille Examinateur Emmanuel Tertre Professeur, Université de Poitiers
Examinatrice Sabine Valange Maître de conférences-HDR, Université de Poitiers Directrice de thèse Sabine Petit Directrice de recherche CNRS, Université de Poitiers
1 « Il est vrai que la nature commence par le raisonnement et finit par l’expérience ;
qu’importe ν il faut prendre la route opposée, commencer par l’expérience et faire en sorte, par
son moyen, d’en découvrir la raison »
3 Voici venu le temps des remerciements après ces trois années de thèse passées à Poitiers. Je remercie toutes les personnes qui ont contribué directement ou indirectement (notamment lors des pauses café) à cette thèse qui, sans eux, n’aurait pas été si intellectuellement et humainement passionnante.
Tout d’abord merci à Sabine Petit qui m’a permis il y a de ça quelques années de
découvrir le monde de la recherche mais surtout le monde de la synthèse minérale avec un stage de L3. Merci à toi Sabine tout d’abord de m’avoir choisi et ensuite fait confiance pour
mener à bien cette thèse et finalement de m’avoir transmis une partie de ton savoir et de ta
rigueur scientifique.
Je tiens également à remercier mes encadrants de stage de Master : Patricia Patrier et Daniel Beaufort, qui ont eu la lourde tâche de former un étudiant. Merci à vous de m’avoir fait confiance !
Ensuite, je tiens à remercier l’ensemble de mon jury de thèse qui a eu l’amabilité de
lire et de juger mes travaux : Antoine Thill et Nicolas Mangold qui ont dû lire l’intégralité de cette thèse pendant leurs vacances d’été (je m’en excuse encore), Emmanuel Tertre qui a bien voulu accepter de présider la soutenance, et ensuite Olivier Grauby et Sabine Valange. Merci à tous pour la discussion riche et passionnante qui a eu lieu, cela a été un réel plaisir malgré les 40°C dans la salle.
Je voudrais exprimer ma gratitude avec une mention très spéciale à Alain Decarreau
qui m’a transmis son savoir immense sur la synthèse d’argiles, et surtout sa passion de la
recherche. Je tiens à souligner que toutes les discussions que nous avons eu ont fait avancer cette thèse ou ont fait ressortir des questions plus que pertinentes. Merci Alain pour tout cela, mais aussi pour votre sympathie et toutes ces discussions.
Merci à l’équipe HydrASA de m’avoir accueilli durant ces 5 belles années : Arnaud
Mazurier, Pierre Chansigaud, Romain Brechon, Philippe Vieillard, Fabien Hubert, Eric Ferrage, Emmanuel Tertre, Claude Fontaine, Claude Laforest, Nathalie Dauger, Benoît Nauleau, Abder El Albani, Dimitri Pret, Paul Sardini, Laurent Caner, Alain Meunier, Thierry Bonifait, Céline Boissard, Stephan Hedan, Gilles Porel, et Philippe Cozensa.
Je tiens également à remercier les filles du service commun de l’IC2MP, Christine Canaff, Julie Rousseau, Nadia Guignard, Christelle Roudaut et Sophie Morisset de la plateforme Prémicat pour leur grande gentillesse et disponibilité mais surtout pour leur aide
dans la partie analytique. J’ai vraiment beaucoup apprécié de travailler avec vous toutes.
Un grand merci à tous les thésards, post-docs et maintenant amis qui ont permis de rendre cette thèse vivante et bien fendard : Valentin Robin, Jean-Christophe Viennet, Liva Dzene, Thomas Riegler, Sophie Billon, Anne-Laure Fauchille, Benoît Merckx, Benoît Hebert, Axel Angileri, Charli Delayre, Olabode Bankole, Nathaelle Onanga Mavotchy, Genia Soldatenko, Natalia Matskova, Carmen Ciotonea, Tojo Radimy, le paléontologue : Florian
4 Bisous !
Un détour par le soleil et le Brésil, pour remercier les collègues brésiliens qui ont passé du temps au labo et en soirée : Obrigado a Edson Bertoluzzi, Thalès Tiecher, Vanessa Bertolazi, Eliana Mano et Jackson Korchagin.
Une pensée amicale et surtout un grand merci aux « petits » stagiaires que j’ai co-encadré durant ces trois années dont deux sont maintenant thésards : Maximillien Mathian et Eloi Le Floch, mais aussi aux autres étudiants : Jason Maurice, Karl Lelièvre, et Gaël Cherfallot.
Merci à mes collocs du bureau 319a qui se sont succédés durant ces trois années : Sylvain Bruzac et Marc Reinholdt pour avoir supporté (et le mot est très faible) mes discussions existentielles et autres questions débilles que je débitais à un rythme soutenu.
Une thèse ne peut bien se dérouler que s’il y a une flopée de potes (je ne vais pas les citer pour ne pas en oublier) qui permettent de se changer les idées au travers de « quelques » soirées mémorables dont la plupart accompagnées d’un gros trou noir…
Pour finir, un grand merci à toute ma famille et belle-famille qui m’ont toujours encouragé et surtout supporté durant ces trois années. Je voudrais vraiment remercier mes parents qui m’ont toujours laissé faire ce que je voulais de ma vie ce qui m’a permis d’arriver
jusqu’à la thèse. Je tiens à remercier chaleureusement Catherine Verron pour les corrections d’orthographe, particulièrement sur les parties de texte écrites entre 1h et 4h du matin. Un très
grand merci à Héloïse pour son soutien sans faille même dans les plus durs moments. Merci à tous
7
Table des matières
Introduction ... 11
Chapitre 1 : Rappels généraux ... 15
1. Structure des phyllosilicates ... 15
1.1. La couche tétraédrique ... 15
1.2. La couche octaédrique ... 15
1.3. L’agencement des couches tétraédriques et octaédriques ... 16
1.4. Substitutions et solutions solides ... 18
1.5. La couche interfoliaire ... 19
1.6. Les phyllosilicates 2:1 : les smectites ... 19
1.6.1. L’empilement des feuillets ... 20
1.6.2. Le gonflement ... 20
1.6.3. L’interstratification ... 20
2. La nontronite ... 21
2.1. Historique ... 21
2.2. Occurrences naturelles ... 21
2.2.1. Altération supergène et sol ... 21
2.2.2. Sédimentaire et hydrothermale ... 21
2.2.3. Magmatisme ... 22
2.3. Structure cristalline et caractéristiques ... 22
2.3.1. Chimie et solutions-solides ... 22
2.3.2. Morphologie ... 23
2.3.3. Caractéristiques ... 24
2.4. Synthèses hydrothermales antérieures ... 25
Chapitre 2 : Protocoles expérimentaux ... 29
1. Synthèses hydrothermales ... 29
1.1. Synthèses à partir d’un co-précipité séché ... 29
1.2. Synthèses à partir d’un co-précipité hydraté ... 31
1.3. Synthèses hydrothermales en milieu réducteur ... 32
1.4. Saturation des échantillons avec Ca2+ ... 33
8
2.1. Spectroscopie infrarouge à transformée de Fourier (FTIR) ... 34
2.1.1 FTIR dans le domaine moyen IR (400 – 4000 cm-1) ... 34
2.1.2 FTIR dans le domaine proche IR (4000 – 10 000 cm-1) ... 34
2.2. Spectroscopie Mössbauer ... 34
2.3. Spectroscopie photoélectronique X (XPS) ... 35
2.4. Diffraction de rayons X (DRX) ... 35
2.5. Analyses chimiques par microscopie électronique à balayage (MEB) associée à un spectromètre à dispersion d’énergie (EDS) ... 35
2.6. Microscopie électronique à transmission (MET) associée à un spectromètre à dispersion d’énergie (EDS) ... 36
2.7 Mesure du pH ... 36
2.8 Mesure du Si (aq) total ... 36
2.9 Mesure du Na (aq) et Fe (aq) total ... 36
Chapitre 3 : Lien entre les propriétés du milieu réactionnel et la
cristallochimie des nontronites ... 37
1. Introduction ... 37
2. Synthèse d’une série de nontronites avec une large gamme de teneurs
en Fe(III) tétraédrique ... 38
3. Résultats complémentaires ... 57
4. Discussion ... 59
Chapitre 4 : Signatures spectrales du Fe(III) dans les nontronites
... 61
1. Introduction ... 61
2. Spectroscopie infrarouge ... 62
3. Spectroscopie Mössbauer ... 68
4. Spectroscopie XPS ... 93
5. Discussion ... 96
Chapitre 5 : Influence des paramètres de synthèse ... 101
1. Introduction ... 101
2. Effet de la durée sur la synthèse de nontronite ... 102
3.
Influence du séchage et de l’agrégation ... 119
9
3.2. Synthèse de la série de nontronite sans séchage ... 131
4. Synthèse en milieu potassique ... 138
5. Synthèse en milieu anaérobie de smectites riches en fer ... 141
6. Discussion ... 143
Conclusion & Perspectives ... 147
Références ... 151
Liste des figures ... 167
Liste des tables ... 173
Travaux associés ... 175
1. Développement des synthèses par voie micro-ondes ... 175
2. Études spectroscopiques de séries chimiques de minéraux synthétiques
... 183
2.1. Interprétation des spectres infrarouge de la serie lizardite-nepouite ... 183
2.2. Interprétation des signatures spectrales de smectites dioctaédriques Ga-Fe(III) ... 191
2.3. Signature spectrale de la série coffinite-uronothorite pour une application dans le domaine minier ... 205
11
Introduction
Le fer est un élément majeur en terme d’abondance dans la croûte terrestre. Ses propriétés physico-chimiques et sa présence dans de nombreux minéraux de la surface de la Terre, lui permettent de jouer un rôle majeur dans un grand nombre de processus biochimiques et géochimiques fondamentaux. Notamment, l’aptitude du fer à pouvoir
échanger ses électrons rend cet élément incontournable dans tous les processus
d’oxydo-réduction. Ces réactions d’oxydo-réduction jouent un rôle majeur dans la mobilité de certains métaux dans les systèmes géologiques, la maturation ou la dégradation de la matière organique, mais encore sur la corrosion et la biocorrosion des métaux. En effet, le cycle biogéochimique du fer présente un grand nombre d’interconnections dans l’environnement entre les phases minérales porteuses de fer et les phases organiques ou biologiques (Murad & Fischer, 1988). Un grand nombre de minéraux sont porteurs de fer au sein de leur structure (minéraux mafiques, sulfures, carbonates, (oxy)(hydr)oxydes, minéraux argileux…). Certains de ces minéraux, notamment les (oxy)(hydr)oxydes de fer et les minéraux argileux jouent un rôle majeur dans les processus biogéochimiques (Stucki, 2013).
Les minéraux argileux, et plus particulièrement les minéraux argileux gonflants tels que les smectites, sont ubiquistes à la surface de la Terre. Ces smectites, de par leur grande
capacité d’échange cationique et leur grande surface spécifique, sont au centre des réactions
biogéochimiques ayant place dans les environnements de surface. La particularité de ces
minéraux gonflants est l’incorporation en grande quantité dans leur structure aussi bien du fer
ferreux Fe(II) que du fer ferrique Fe(III). Cette particularité permet des échanges d’électrons entre le fer structural des smectites et leur environnement tout en conservant le fer dans leur structure. Les propriétés du fer structural dans les smectites ainsi que l’impact des processus
d’oxydo-réduction sur les propriétés intrinsèques des smectites ont fait l’objet de nombreuses
études (voir revues bibliographiques, Stucki et al., 1988; Stucki, 2011, 2013).
Des smectites riches en fer ont également été identifiées à la surface de Mars à partir des données hyperspectrales (e.g. Poulet et al., 2005; Bibring et al., 2006; Loizeau et al., 2007; Mustard et al., 2008; Bishop et al., 2008; Carter et al., 2010). L’identification de ces minéraux par spectroscopie infrarouge dans le domaine proche infrarouge (NIR) est réalisée par comparaison des spectres obtenus par imagerie hyperspectrale avec des spectres de minéraux argileux de référence. Dans ce contexte, un regain d’intérêt a été observé pour
l’obtention de données spectroscopiques robustes et appropriées pour l’identification des
signatures spectroscopiques des minéraux argileux. La découverte de minéraux argileux sur Mars met en avant la nécessité de cerner les conditions de formation de ces minéraux. Ces problématiques placent la synthèse de minéraux argileux comme un outil indispensable pour fournir des minéraux de référence avec une chimie contrôlée, et obtenir les signatures spectrales de ces minéraux, tout en cernant leurs conditions de formation (Andrieux & Petit, 2010; Chemtob et al., 2015; Petit et al., 2015).
D’un point de vue applicatif, ces smectites riches en fer sont largement utilisées
comme précurseurs pour l’élaboration de matériaux. Ceux-ci sont ensuite utilisés dans
12
et al., 2015 / Travaux associés §3), la sorption de polluants (e.g. Gupta et al., 2006), la
dégradation de composés organiques (e.g. Hofstetter et al., 2006; Neumann et al., 2009),
l’immobilisation de métaux lourds (e.g. Jaisi et al., 2009; Yang et al., 2012; Ilgen et al., 2012) ou encore les cristaux liquides (e.g. Michot et al., 2008, 2009, 2013; Paineau et al., 2013; MacGregor-Ramiasa et al., 2015).
Malgré de nombreuses études menées sur les smectites ferrifères, des caractérisations fondamentales restent difficiles à réaliser, comme la distribution du fer dans leur structure. Si le Fe(II) est bien connu pour être présent uniquement dans la couche octaédrique de ces smectites, le Fe(III), quant à lui, peut être présent aussi bien dans la couche octaédrique que dans les couches tétraédriques. Il est bien admis dans la littérature que le Fe(III) est essentiellement présent dans les sites octaédriques des smectites. Dans le cas de smectites dioctaédriques riches en Fe(III) telles que les nontronites, l’existence de Fe(III) en sites tétraédriques ([4]Fe(III)) est cependant inévitable. Néanmoins, le [4]Fe(III) est difficile à mettre en évidence et à quantifier. Ces difficultés sont, par exemple, illustrées par la grande diversité des résultats de quantification du [4]Fe(III) dans la nontronite de Garfield (Osthaus, 1954; Goodman et al., 1976; Rozenson & Heller-Kallai, 1977; Bonnin et al., 1985; Johnston & Cardile, 1985; Cardile, 1989; Luca, 1991).
Dans ce contexte, l’étude détaillée des signatures spectrales du Fe(III) dans les nontronites est requise afin d’identifier et quantifier le Fe(III) octaédrique ([6]Fe(III)) et le [4]Fe(III) dans le but par exemple, d’appréhender les mécanismes de transfert d’électrons
impliquant le fer structural. Notamment, la réactivité des smectites au processus d’oxydo-réduction est bien connue pour être très sensible à leur cristallochimie (Neumann et al., 2008, 2009, 2011), mais le rôle joué par le [4]Fe(III) dans les transferts d’électrons est très peu compris.
Les nontronites naturelles présentent la plupart du temps une composition chimique complexe du fait de leur origine et leur mode de formation (elles sont principalement décrites comme des produits d’altération de minéraux primaires). De plus, ces minéraux argileux naturels se retrouvent très souvent dans des associations complexes de minéraux. Cette complexité chimique et minéralogique rend difficile l’interprétation des données spectroscopiques. L’étude détaillée des signatures spectrales du Fe(III) se trouve grandement facilitée par l’emploi d’une approche par synthèses minérales afin d’avoir des échantillons monominéraux avec une chimie simple et contrôlée.
Le travail présenté ici a tout d’abord porté sur l’élaboration d’une série de nontronites synthétiques présentant une gamme de teneur en [4]Fe(III) la plus large possible. Une revue des synthèses antérieures de nontronite a été réalisée afin de déterminer les paramètres optimaux de synthèse tout en identifiant le ou les paramètres pouvant entraîner une variation des teneurs en [4]Fe(III) des nontronites. L’élaboration de la série dans des conditions
contrôlées a permis de mettre en évidence certains facteurs/contraintes chimiques pouvant influencer la formation de la nontronite et la teneur en [4]Fe(III) des nontronites dans un système extrêmement simplifié (Si-Fe(III)-Na). Une étude des signatures spectroscopiques du Fe(III) dans les smectites a été réalisée par spectroscopies infrarouge (IR), Mössbauer, et photoélectronique X (XPS). Une étude cinétique a ensuite été menée afin d’étudier la stabilité de ces nontronites ferrifères dans les conditions expérimentales et de suivre leur évolution au cours du temps. Elle a permis d’affiner la méthodologie de synthèse et d’apporter une
13 meilleure compréhension du processus de synthèse mis en jeu dans cette thèse. Ainsi les relations nontronite ferrifère - aegirine ont pu également être affinées. Enfin, dans une dernière partie sont regroupés les travaux associés auxquels j’ai participé durant ma thèse. Ils concernent le développement de synthèses par voie micro-ondes, l’acquisition de données spectroscopiques à partir de séries de minéraux synthétiques et l’application de smectites synthétiques riches en fer pour l’élaboration de matériaux catalytiques pour le traitement
15
Chapitre 1 : Rappels généraux
1. Structure des phyllosilicates
Les phyllosilicates sont des minéraux lamellaires de petite taille, formés de feuillets constitués de couches de polyèdres élémentaires (octaèdre et tétraèdre). La structure de ces polyèdres est déterminée par la coordinance des cations qui les composent.
1.1. La couche tétraédrique
La couche tétraédrique est composée de tétraèdres de SiO44- ou Al(III)O45- et parfois
Fe(III)O45- (Figure 1a, b). Ces tétraèdres sont reliés entre eux par le partage des trois oxygènes
basaux (Figure 1c, d, e). Chaque anion O2- dans le plan basal est donc lié à deux cations Si(IV) ou un Si(IV) et un Al(III)/Fe(III), puisque deux cations trivalents ne peuvent être voisins au sein de la couche tétraédrique (règle de Löwenstein). Les tétraèdres forment un réseau bidimensionnel où les oxygènes apicaux sont tous situés du même côté du plan basal. Le réseau de tétraèdres délimite des cavités hexagonales (Figure 1c).
Figure 1: Représentation d’un tétraèdre (a) en vue éclatée (b) sous forme de polyèdres et agencement de ces tétraèdres au
sein d’une couche tétraédrique vue suivant l’axe c (c), l’axe a (d) et l’axe b (e).
1.2. La couche octaédrique
La couche octaédrique est constituée d’octaèdres, couchés sur une de leur face
triangulaire et reliés entre eux par leurs arêtes et donc leurs 6 oxygènes (Figure 2a, b, c, d, e). Les octaèdres sont formés par des cations (R) divalents (R(II)O610-) ou trivalents (R(III)O69-).
Deux types de réseaux existent selon que la couche octaédrique est composée principalement de cations divalents ou trivalents. Le type trioctaédrique correspond à l’occupation de trois sites octaédriques adjacents, où chaque anion O2- est lié à trois cations divalents (Figure 2g). Le type dioctaédrique correspond à l’occupation de deux sites octaédriques adjacents sur trois, le troisième site étant vacant (lacune), et où chaque anion O2- est lié à deux cations trivalents (Figure 2f). a b c d e a b c b a c b cavité hexagonale
16
Figure 2: Représentation d’un octaèdre (a) en vue éclatée (b) sous forme de polyèdres et agencement de ces octaèdres au
sein d’une couche octaédrique vue suivant l’axe c (c), l’axe a (d) et l’axe b (e). Représentation polyédrale d’une couche
dioctaédrique (f) et trioctaédrique (g).
1.3. L’agencement des couches tétraédriques et octaédriques
L’accrochage de la couche tétraédrique avec la couche octaédrique s’effectue par la
mise en commun des oxygènes apicaux des tétraèdres avec les octaèdres (Figure 3d). Néanmoins, les dimensions cristallographiques théoriques de ces deux types de couches sont
différentes. Par conséquent, l’ajustement des couches octaédrique et tétraédrique ne peut se
réaliser sans déformations. La couche tétraédrique se déforme par rotations des tétraèdres suivant des axes perpendiculaires et parallèles au plan basal (Figure 3a, b). Les déformations de la couche octaédrique sont relativement faibles dans le cas d’une structure trioctaédrique, alors que pour une structure dioctaédrique, la présence de sites vacants permet la déformation des octaèdres occupés (Figure 3c).
a b c d e a b c b c a a f g
17
Figure 3 : Déformations de la couche tétraédrique avec basculement (a) et rotation (b) des tétraèdres, déformation de la couche octaédrique (c), et agencement et accroche des couches octaédriques et tétraédriques (d).
Figure 4 : Représentation d’un feuillet de type 1 :1 et de type 2 :1.
Les phyllosilicates peuvent être décrits avec deux types de structure cristalline : le type 1:1 et 2:1 (Figure 4). Le type 1:1 est celui au sein duquel un feuillet est formé d’une couche octaédrique liée à une couche tétraédrique. Pour ce type de feuillet, les 6 sommets d’un site octaédrique sont composés par 4 groupements OH- et 2 oxygènes mis en commun avec les tétraèdres. Le type 2μ1 regroupe les feuillets composés d’une couche octaédrique liée de part
et d’autre à deux couches tétraédriques. Pour ce type de feuillets, les 6 sommets de chaque
site octaédrique se composent de 2 groupements OH- et 4 oxygènes mis en commun avec les tétraèdres des deux couches tétraédriques. La couche octaédrique de ces feuillets 2:1 est formée de deux types d’octaèdres : deux tiers sont des octaèdres cis et l’autre tiers des
octaèdres trans. Les deux types se différencient par la localité des groupements OH-. Ces
a b
c d
18 groupements sont situés sur un côté d’un octaèdre pour le type cis, ou sur la grande diagonale
d’un octaèdre pour le type trans (Figure 5).
Figure 5 : Configuration d’une couche dioctaédrique cis-vacante (a), (b) et trans-vacante (c).
1.4. Substitutions et solutions solides
Au sein des feuillets, un grand nombre de substitutions cationiques peut intervenir dans les couches tétraédriques et octaédriques. Ces substitutions peuvent être homovalentes -
c’est-à-dire qu’un cation est remplacé par un autre ayant la même valence, ou hétérovalente -
ce qui correspond au remplacement d’un cation par un autre ayant une valence différente. Une solution solide peut être formée entre deux espèces minérales (pôles de la solution solide) si, grâce au jeu des substitutions, le passage d’une espèce minérale à l’autre est réalisable. Vegard (1928) montra que les paramètres de maille évoluent de façon linéaire en fonction du nombre de substitutions dans le réseau cristallin. Une solution solide peut être continue ou partielle selon que les espèces minérales sont isostructurales ou non, et si les cations mis en jeu suivent les règles de Goldschmidt ou non. Les solutions solides peuvent être décrites en fonction du mécanisme de la variation chimique pouvant être observé le long
d’une série. Les solutions solides de substitution décrivent le remplacement isomorphe d’un ion par un autre au sein d’un même site cristallographique. L’ajout d’un atome dans un site vacant pour obtenir la neutralité se décrit comme une solution solide d’addition. Et la création d’une vacance se décrit comme une solution solide de soustraction.
Les substitutions octaédriques. Les couches octaédriques peuvent être le lieu de
différents types de substitutions : homovalentes (e.g. Al(III) → Fe(III), Mg(II) → Fe(II)), hétérovalentes (e.g. Al(III) → Mg(II), Fe(III) → Mg(II), Mg(II) → Li(I)), par la création
d’une lacune (e.g. Mg(II) → □).
Les substitutions tétraédriques. Au niveau de la couche tétraédrique, les substitutions
sont, dans la nature, essentiellement hétérovalentes du type Si(IV) → Al(III) et Si(IV) → Fe(III).
Les substitutions hétérovalentes au sein des couches tétraédriques et octaédriques et la création de lacunes dans la couche octaédrique, entraînent une perte de l’électroneutralité du feuillet compte tenu de la différence de valence entre deux cations ou un cation et une lacune.
Cette perte d’électroneutralité entraîne l’apparition d’une charge négative permanente des
feuillets.
OH
cis vacante trans vacante
19
1.5. La couche interfoliaire
L’électroneutralité lorsqu’elle n’est pas assurée - c’est le cas particulièrement dans les
feuillets des phyllosilicates de type 2:1 - est obtenue par des cations compensateurs et leur
sphère d’hydratation (constituée de molécules polaires) se situant entre les feuillets. Ces
cations peuvent parfois se condenser et former une couche octaédrique (sans sommets communs avec les couches tétraédriques des feuillets) plus ou moins continue entre les feuillets. Les substitutions cationiques (Ca2+, Mg2+, K+, Na+…) au sein de la couche interfoliaire sont très courantes et plus variées que celles intervenant dans les couches octaédriques et tétraédriques.
1.6. Les phyllosilicates 2:1 : les smectites
Cette famille de phyllosilicates est subdivisée en différents groupes en fonction de la valeur de la charge négative du feuillet issue des substitutions hétérovalentes et de leur localisation, et de l’état d’hydratation du cation interfoliaire (Table 1).
Charge électrique foliaire et état
d’hydratation du cation interfoliaire Minéraux dioctaédriques Minéraux trioctaédriques
x = 0 pyrophyllite talc
0.3 < x < 0.6 ; cations échangeables hydratés smectites smectites 0.6 < x < 0.9 ; cations échangeables hydratés vermiculites vermiculites
x ≈ 0.9 ; cations déshydratés illite, glauconite
0.9 < x < 1 ; cations déshydratés micas micas 1.8 < x < 2 ; cations déshydratés micas durs
x ≈ variable ; couche hydroxylée chlorites chlorites
Table 1 : Classification simplifiée des phyllosilicates de type 2:1 d’après (Guggenheim et al., 2006).
Le groupe des smectites englobe les smectites dioctaédriques et les smectites trioctaédriques (Table 2). Les smectites ont des charges foliaires très variables issues de substitutions hétérovalentes se produisant dans la couche tétraédrique (Si(IV) → Al(III) et Si(IV) → Fe(III)) et/ou dans la couche octaédrique (Al(III) → Mg(II), Fe(III) → Mg(II), Mg(II) → Li(I), Mg(II) → □). Ces smectites peuvent se classifier suivant : (i) la nature du cation octaédrique majoritaire, (ii) la nature des substitutions hétérovalentes majoritaires et leurs localisations (Table 2).
Localisation de la charge Smectites dioctaédriques Smectites trioctaédriques
octaédrique
montmorillonite hectorite
Si4(Al2-yMgy)O10(OH)2, M+y Si4(Mg3-yLiy)O10(OH)2, M+y
stévensite
Si4(Mg3-y□y)O10(OH)2, M+2y
tétraédrique
beidellite saponite
(Si4-xAlx)Al2O10(OH)2, M+y (Si4-xAlx)Mg3O10(OH)2, M+y
nontronite
(Si4-x Alx)Fe(III)2O10(OH)2, M+y Table 2 : Classification du groupe des smectites.
20
1.6.1. L’empilement des feuillets
Les phyllosilicates 2:1 sont constitués d’un empilement de feuillets selon l’axe c. Ces
feuillets peuvent s’empiler selon trois grands types d’empilement : ordonné, semi-ordonné et
turbostratique (Figure 6). L’empilement ordonné se traduit par des translations régulières à grande échelle entre des feuillets successifs, c’est par exemple le cas des micas. Un empilement semi-ordonné peut être observé pour des phyllosilicates 2:1 ayant une charge négative d’origine tétraédrique, comme par exemple certaines smectites ou vermiculites. Enfin, l’empilement turbostratique correspond à un empilement non-rationnel entre feuillets
successifs, c’est le cas principalement des smectites. Pour ces dernières, la cohérence d’empilement varie en fonction de la nature du cation interfoliaire et son degré d’hydratation,
ainsi que de la localisation et du taux de substitutions hétérovalentes au sein du feuillet (Cases et al., 1992, 1997; Berend et al., 1995).
Figure 6 : Les différents types d’empilement dans les phyllosilicates.
1.6.2. Le gonflement
La propriété de gonflement caractérisant les smectites et les vermiculites peut être déterminée par la capacité des cations interfoliaires à conserver leur sphère de molécules
polaires (molécules d’eau, de glycérol, d’éthylène glycol… ; Sato et al., 1992; Cases et al., 1997). Pour les molécules d’eau, c’est donc l’affinité pour l’eau du cation interfoliaire qui va
gouverner l’état d’hydratation pour une humidité relative donnée (Sato et al., 1992; Berend et al., 1995; Cases et al., 1997; Ferrage et al., 2010, 2011).
Pour un cation donné, les molécules d’eau s’organisent en couches plus ou moins
continues dans l’espace interfoliaire suivant l’humidité relative. Partant de l’état déshydraté (0
couche de molécules d’eau (0W)), l’augmentation de l’humidité relative induit l’adsorption de molécules d’eau selon différentes configurations stables de l’espace interfoliaire dans lequel les molécules d’eau sont intercalées en 1 (1W), 2 (2W), ou 3 (3W) couches (Nagelschmidt,
1936; Norrish, 1954; Walker, 1958; Harward & Brindley, 1965; Harward & Carstea, 1967; Watanabe & Sato, 1988; Sato et al., 1992; Yamada et al., 1994; Tamura et al., 2000; Ferrage
et al., 2007, 2010, 2011; Dazas et al., 2014).
1.6.3. L’interstratification
L’interstratification peut aussi bien décrire l’empilement de feuillets de nature
différente (hétérogénéité de structure) comme par exemple les interstratifiés illite/smectite,
que l’empilement de feuillets de même nature mais avec différents états d’hydratation
empilement
ordonné
empilement
semi-ordonné
empilement
turbostratique
21
(hétérogénéité d’hydratation) comme par exemple les interstratifiés smectite (1W)/smectite
(2W).
2. La nontronite
Dans cette partie, une revue bibliographique succincte des occurrences et des caractéristiques de la nontronite a été réalisée. Pour une revue complète et détaillée, se référer à Wilson (2013).
2.1. Historique
La nontronite a été pour la première fois découverte et décrite près de Nontron (France) par Berthier (1827). Cette nontronite de couleur jaune pâle à jaune vif se présente en nodules millimétriques à centimétriques disséminés dans un minerai de manganèse. Berthier (1827) décrit la nontronite comme : un minéral opaque, de cassure inégale, onctueux au
toucher, très tendre (rayable à l’ongle), qui absorbe de l’eau, très facilement dissoute par de l’acide chlorhydrique et qui est essentiellement composé de silice, de fer et d’eau de
« combinaison ».
2.2. Occurrences naturelles
2.2.1. Altération supergène et sol
La nontronite est, dans la littérature, principalement décrite comme le produit résultant
de l’altération de roches volcaniques basiques (Allen & Scheid, 1946; Sherman et al., 1962; Benson & Teague, 1982; Jakobsson & Moore, 1986; Caillaud et al., 2005; Meunier et al., 2008). Nahon et al., (1982) ont montré la formation de nontronites à partir d’olivines dans un
profil latéritique issu de l’altération de roches ultrabasiques. Néanmoins, l’altération de roches
volcaniques rhyolitiques et sulfurées peut également entraîner la formation de nontronites à partir de la dissolution de la pyrite et des feldspaths alcalins par une solution oxydante (Fernández-Caliani et al., 2004). Les études de l’altération de roches métamorphiques comme des skarns (principalement des pyroxénites) par Eggleton (1975), Decarreau et al. (1987) et Keeling et al. (2000) montrent la formation de nontronites à partir de l’altération des pyroxènes ou parfois des biotites.
Les nontronites sont considérées comme un minéral éphémère lorsqu’elles sont vues
dans l’ensemble de l’altération (Wilson, 1999, 2013). En effet, l’évolution de l’altération tend
vers la déstabilisation de la nontronite et sa décomposition en kaolinite et hématite. Van Ranst & De Coninck (1983) rapportent la formation de nontronites dans l’horizon B des sols glauconitiques à partir de l’oxydation du Fe(II) présent dans les glauconites et du lessivage du potassium. La formation de nontronites est également observée dans l’horizon C d’un vertisol dérivé de l’altération d’un basalte de Sardaigne (Vingiani et al., 2004). Des smectites néoformées riches en Fe(III) ont également été observées dans de nombreuses études sur la minéralogie des sols (Kantor & Schwertmann, 1974; Bui & Wilding, 1988; Pai et al., 1999; Ryan & Huertas, 2009)
2.2.2. Sédimentaire et hydrothermale
La nontronite est commune dans les sédiments océaniques associés à une activité hydrothermale des fonds marins (Bischoff, 1972; Benson & Teague, 1982; Cole & Shaw,
22 1983; McMurtry et al., 1983; Singer et al., 1984; Badaut et al., 1985, 1992; Jakobsson & Moore, 1986; Alt, 1988; Decarreau et al., 1990; Kohler et al., 1994; Ueshima & Tazaki, 2001; Severmann et al., 2004; Dekov et al., 2007). La nontronite est un minéral authigène dans ces environnements qui peut se former soit par l’altération de produit volcanique, soit directement ou indirectement à partir des fluides hydrothermaux relargués par l’activité hydrothermale des fonds marins. Les nontronites sont principalement formées à partir de saumures chaudes et métalliques dans différents environnements de température et de condition oxydo-réductrice.
Une origine purement sédimentaire de la nontronite a aussi été avancée dans des conditions oxydantes à la température des fonds marins (2 - 3°C) à partir de la transformation
d’hydroxyde de fer et de silice biologique (Cole, 1985).
Keeling et al., (2000) ont montré que la nontronite peut être le produit d’altération liée à un évènement hydrothermal de basse température associé à de la fracturation dans des roches métamorphiques (schistes, gneiss, amphibolite).
2.2.3. Magmatisme
Oyawoye & Hirst (1964) ont été les premiers, à notre connaissance, à proposer une origine « primaire » pour des smectites nontronitiques dans les granites du Nigéria. Ces auteurs ont observé dans la roche fraîche la présence de nontronites entourées de cristaux de quartz euhédriques, de feldspaths et de biotites. Aucune figure de dissolution des cristaux de biotites, feldspaths et quartz n’a été observée dans ces roches. De plus, la nontronite n’est jamais observée sur les parois des cavités ou dans les joints de grains, indiquant une origine primaire. Ces auteurs suggèrent donc une formation de la nontronite à la fin de la cristallisation du magma à partir des fluides magmatiques résiduels de haute température (supérieure à la température de fluides hydrothermaux). Une interprétation similaire a été décrite par Meunier et al. (2008, 2012) sur la formation de nontronites à partir de fluides en ébullition peu de temps après la mise en place de roches basaltiques, ou de fluides magmatiques résiduels. Berger et al. (2014) montrent également par des observations pétrographiques la formation de minéraux argileux riches en Fe à partir de fluides magmatiques acides, de haute température, liés au dégazage de la coulée basaltique à la fin de
l’épisode éruptif.
2.3. Structure cristalline et caractéristiques
La nontronite est un phyllosilicate dioctaédrique de type 2:1 (Figure 7). Méring & Oberlin (1971), Besson et al. (1983) et Tsipursky & Drits (1984) ont déterminé par diffraction électronique une symétrie hexagonale centrosymétrique indiquant une occupation trans-vacante de la couche octaédrique.
Le calcul du diffractogramme de la nontronite effectué par Méring & Oberlin (1971) a
démontré l’existence d’une rotation des tétraèdres et d’une distorsion des octaèdres afin de
représenter au mieux les intensités observées. L’étude combinée de diffraction de rayons-X et de diffraction électronique de Méring & Oberlin (1971) a mis en évidence un empilement désordonné turbostratique.
2.3.1. Chimie et solutions-solides
La composition chimique idéale de la couche octaédrique d’une nontronite est
23 homovalentes et hétérovalentes peuvent se produire dans la couche octaédrique notamment avec Al(III), Mg(II), Fe(II) ou encore Ni(II) (e.g. McMurtry et al., 1983; Petit et al., 1992, 2015; Grauby et al., 1994; Köster et al., 1999; Gaudin et al., 2004, 2005; Andrieux & Petit, 2010; Mano et al., 2014). Les couches tétraédriques quant à elles sont constituées de cations Si(IV) pouvant être substitués par des cations Al(III) et/ou Fe(III) (e.g. Goodman et al., 1976; Manceau et al., 2000b; Andrieux & Petit, 2010; Decarreau & Petit, 2014; Petit et al., 2015). La charge liée aux substitutions hétérovalentes tétraédriques et octaédriques est compensée
par l’incorporation de cations compensateurs dans l’espace interfoliaire. La nontronite est
considérée comme le pôle Fe(III) du groupe des smectites dioctaédriques (Table 2). Newman & Brown, (1987) définissent toutes smectites ayant une teneur en Fe(III) octaédrique > 1.5 atomes par O10(OH)2 comme étant des nontronites.
Des solutions-solides ont été mises en évidence entre nontronites et montmorillonites riches en Fe(III) (McMurtry et al., 1983; Petit et al., 1992; Köster et al., 1999; Gaudin et al., 2004, 2005). Expérimentalement, par synthèses minérales, des séries chimiques ont également été mises en évidence entre nontronites et saponites (Grauby et al., 1994), entre nontronites et beidellites (Andrieux & Petit, 2010; Petit et al., 2015) et entre nontronites et smectites-Ga(III) (Petit et al., 2016 / Travaux associés §2.2).
Figure 7 : Structure de la nontronite d’après les données de Manceau et al. (1998).
2.3.2. Morphologie
Les nontronites naturelles ont le plus souvent une morphologie en latte de quelques centaines de nanomètres à quelques microns de longueur (Mackenzie et al., 1952; Méring & Oberlin, 1971). Ces particules de quelques feuillets d’épaisseur peuvent être libres ou c
b
24
s’empiler de façon turbostratique pour former des agrégats. Ces particules en latte tendent parfois à s’associer bout à bout formant des rubans épais et plissés (Méring & Oberlin, 1971). D’autres types de morphologie plus rares ont été observés, comme des sphères (Güven, 1988),
des fibres (Kurnosov et al., 547; Cole & Shaw, 1983; Singer et al., 1984), ou encore des feuillets repliés ou roulés (Kohler et al., 1994; Taitel-Goldman & Singer, 2001).
2.3.3. Caractéristiques
Echange cationique et surface spécifique. La charge foliaire de la nontronite étant
principalement localisée au sein de la couche tétraédrique, les caractéristiques d’échange cationique sont similaires à celles de la beidellite. La capacité d’échange cationique (CEC) de
la nontronite a été mesurée entre 95 et 110 meq/100g à l’aide de différentes méthodes par
Czímerová et al. (2006). Perronnet et al. (2007) ont déterminé les surfaces basales et de bords de la nontronite de Garfield par adsorption d’azote (N2), avec une surface basale de 50.3 m².g -1 et une surface de bordure de 27.2 m².g-1. Decarreau et al. (2014) ont déterminé par adsorption d’argon à basse pression les surfaces basales et de bordures de nontronites
synthétisées entre 75 et 150°C. Ces auteurs ont obtenu une surface basale entre 182 et 143 m².g-1 et une surface de bordure entre 155 et 77 m².g-1 avec l’augmentation de la température
de synthèse.
Couleur. La nontronite peut se présenter sous différentes teintes de couleur : marron,
marron-vert, marron-jaune, jaune-olive, vert-olive, jaune-vert et vert. Keeling et al. (2000) ont observé un changement de couleur de la nontronite de la mine d’Uley (Nau-2) lorsque la température diminuait. La couleur initiale marron de la nontronite est passée à marron-vert vers -75°C puis jaune-vert vers -125°C jusqu’à vert à -175°C. Ce changement de couleur est probablement lié à des arrangements structuraux et aux proportions relatives du Fe(III) octaédrique et tétraédrique (Gates et al., 2002). Un changement de couleur a également été
observé lorsqu’une nontronite initialement de couleur jaune-olive est soumise à des impacts
de 180 kbars (marron-jaune) et 300 kbars (marron) (Weldon et al., 1982). Dans ce cas, le changement de couleur pourrait être lié à une diminution de la cristallinité, autrement dit une augmentation du désordre des sites cristallographiques de la nontronite lors de l’impact comme décrit par Friedlander et al. (2015). Lors de réductions microbiennes ou chimiques de la nontronite, des changements de couleur ont été observés, partant d’une couleur marron-jaune (oxydé) et allant jusqu’au bleu-noir (réduit) (e.g. Koo et al., 2014).
Dissolution. La nontronite est très facilement dissoute par des acides forts tels que
l’acide chlorhydrique ou l’acide sulfurique. Cependant, les mécanismes de dissolution de la
nontronite sont encore mal connus. Luca & MacLachlan (1992) ont observé, lors du traitement acide (HCl 10%) d’une nontronite (NG-1), que le Fe(III) octaédrique et le Fe(III) tétraédrique sont extraits de la structure cristalline à la même vitesse. Gainey et al. (2014) ont
observé une dissolution stœchiométrique de la nontronite (Nau-1) à pH < 1 (HCl) et non-stœchiométrique à pH > 1.
Une étude au microscope à force atomique (AFM) de la dissolution de la nontronite de Garfield à pH 1.5 – 2 (HCl) montre que les cristaux se dissolvent à partir de leur bords alors que les surfaces basales restent intactes (Bickmore et al., 2001). Ces auteurs ont aussi observé
25 une dissolution préférentielle de certains sites de bordure. En effet, les faces cristallines ayant une forte affinité à la protonation (i.e. des faces étant protonées dont la charge négative des
oxygènes n’étant pas compensée) se dissolvent plus vite.
Thermique. La déshydratation et la déhydroxylation d’une nontronite saturée Ca2+ ont
été étudiées en détail par Frost et al. (2000) à l’aide d’analyses thermogravimétriques. La déshydratation de la nontronite entraîne une perte de 12 à 15 % massique entre 60 et 120°C. La déshydratation du cation interfoliaire se produit entre 120 et 160°C, entrainant une perte de masse de 2.5 à 4 %. Au-dessus de 160°C, une perte de masse d’environ 1 % marque la déshydroxylation des feuillets de nontronite.
L’analyse thermique différentielle de la nontronite montre un endotherme intense à 60
- 120°C lié à la déshydratation et un faible endotherme à environ 350 - 500°C lié à la déshydroxylation. Luca (1991) a montré que la stabilité thermique de la nontronite est dépendante de la nature du cation interfoliaire. La nontronite comme la beidellite possède le plus souvent une température de déshydroxylation plus faible que celle de la montmorillonite.
Cette différence peut être expliquée par la prédominance d’une nature trans-vacante (tv) de la couche octaédrique (Drits et al., 1995; Muller et al., 2000). A une température supérieure à 600°C la nontronite est complètement déshydroxylée et se transforme en hématite (Heller-Kallai & Rozenson, 1980).
Réduction. La nontronite possède la particularité d’être sensible à l’oxydation et à la
réduction suivant les conditions du milieu. Stucki (2011, 2013) propose des revues
bibliographiques complètes de l’implication du fer dans les sols et les minéraux argileux à l’origine de certains processus naturels. Ces revues résument les principaux mécanismes de la
réduction de la nontronite soumise à un changement des conditions oxydo-réductrices du
milieu ou à l’intervention de certains microorganismes, et la répercussion qu’entraîne cette
réduction sur les propriétés physico-chimiques des nontronites. Le mécanisme exact de la réduction est encore soumis à diverses questions, néanmoins Lear & Stucki (1987) et Komadel et al. (1990) proposent que la réduction se produise suivant les surfaces basales et non par les bords des particules. Cette réduction du Fe(III) en Fe(II) entraîne une déshydroxylation des feuillets de nontronite et une migration de certains atomes de Fe de la position cis à une position trans, créant ainsi des domaines trioctaédriques (Stucki & Roth, 1976; Drits & Manceau, 2000; Manceau et al., 2000a; Fialips et al., 2002; Neumann et al., 2011).
2.4. Synthèses hydrothermales antérieures
La synthèse minérale reste aujourd’hui un domaine stratégique dans différentes
sphères scientifiques et industrielles (cosmétique, métallurgie, pharmaceutique…). L’intérêt de la synthèse minérale est double, elle permet d’éclaircir le mode de formation des minéraux et leurs conditions de genèse (température, propriétés physico-chimiques) tout en contrôlant la composition chimique du minéral. Le contrôle de la chimie du minéral permet de distinguer et de déterminer les possibilités de solutions-solides, et d’élaborer les pôles théoriques rares et
26 Les premiers essais de synthèses hydrothermales de nontronites ont été effectués à 350°C pendant une durée de 6 jours par Ewell & Insley (1935). Ces synthèses ont été réalisées à partir d’un gel (Fe2Si2O5) formé par co-précipitation de chlorure ferrique et de
silicate de sodium, puis séché à 110°C. Les auteurs ont cependant identifié un mélange
d’hématite, de nontronite et d’un silicate ferrique hydraté. Hamilton & Furtwängler (1951) ont
synthétisé des nontronites mal cristallisées entre 200 et 400°C à partir de solutions diluées de silicate de sodium et de chlorure ferrique. La formule structurale Si4Fe(III)1.89O10(OH)2Na0.33
de ces produits de synthèse indique plutôt un minéral de type hisingérite (proto-nontronite). Suite à ces synthèses à haute température, des nontronites peu organisées ont été synthétisées dans des conditions anoxiques à température ambiante à partir de solutions de Si (aq) et Fe2+(aq)
par Harder (1976). Cet auteur conclut que la formation de smectite dioctaédrique semble très difficile à réaliser à basse température sans la présence de Mg(II) et Fe(II). Decarreau & Bonnin (1986) ont réussi à synthétiser à 75°C une nontronite de très faible charge par
oxydation d’une smectite ferreuse obtenue dans des conditions réductrices à partir d’un gel
co-précipité selon la méthode décrite par Decarreau (1980). Ce gel est obtenu dans des conditions anoxiques entre des solutions de métasilicate de sodium et sulfate ferreux en présence d’acide sulfurique. Par la suite, des nontronites faiblement cristallisées ont été synthétisées par Decarreau et al. (1987) entre 100 et 150°C dans des conditions oxydantes à partir de la co-précipitation d’un gel entre des solutions de chlorure ferrique et de silicate de sodium en présence d’acide chlorhydrique. Ces nontronites saturées Ca2+ et faiblement cristallisées ont une formule structurale Si4Fe(III)1.83O10(OH)2Ca0.26 similaire à celles
obtenues par Hamilton & Furtwängler (1951). Des smectites Fe(III)/Fe(II) semblables à des nontronites ont été synthétisées par Mizutani et al. (1991) à 150°C dans des conditions anoxiques (dithionite de sodium) à partir de solutions de sulfate ferreux, d’acide silicique et
d’hydroxyde de sodium.
A partir des résultats de ces premières synthèses, il apparait donc que l’obtention du
pôle nontronite purement Fe(III) est très difficile et délicate. En effet, la synthèse en milieu oxydant amène à la formation de minéraux très faiblement cristallisés de type hisingérite. Les seules synthèses de nontronites ont été réalisées en milieu réducteur et requièrent la présence de Fe(II) ou de Mg(II).
Grauby (1993) a obtenu dans le meilleur des cas pour le système purement Fe(III), des nontronites très peu cristallisées similaires à une hisingérite. Ces synthèses ont été réalisées à
200°C pendant une durée de 10 jours à partir d’un gel précurseur séché. Ce gel a été obtenu, d’après la méthodologie établie par Decarreau (1980), par co-précipitation de solutions de
métasilicate de sodium et de chlorure ferrique en présence d’acide chlorhydrique selon la relation suivante :
4 SiNa2O3 + 2 Fe(III)Cl3 + 2 HCl→ Si4Fe(III)2O11 + 8 NaCl + H2O (eq. 1)
Le gel est ensuite séché à 70°C puis broyé avant le traitement hydrothermal.
L’investigation de la solution-solide Fe(III)-Al (Grauby, 1993) a mis en évidence la difficulté d’obtenir des smectites de composition proche du pôle nontronite, mais ces travaux
ont néanmoins montré l’influence déterminante du pH lors de la synthèse sur la nature des minéraux obtenus. Notamment, un pH basique est requis pour l’obtention de smectites.
27 La solution-solide nontronite-saponite a été obtenue par Grauby et al. (1994) à 200°C
à partir d’un gel séché obtenu par co-précipitation entre des solutions de chlorure ferrique
et/ou de magnésium et de silicate de sodium en présence d’acide chlorhydrique.
Au vu des résultats peu probants des synthèses de nontronites purement Fe(III) obtenus par Grauby (1993), des travaux de DEA à l’Université de Poitiers ont porté sur le rôle du pH lors de la co-précipitation du gel et lors de la synthèse (Lecomte, 1998). Les résultats montrent que l’ajout d’acide chlorhydrique lors de la co-précipitation du gel précurseur couplé à un pH non-contraint lors de la synthèse, entraîne la formation d’hématite et de silice amorphe quelle que soit la température (de 180 à 240°C). Lecomte (1998) a réussi à synthétiser une nontronite purement Fe(III) à 180°C en fixant un pH basique lors de la synthèse et la co-précipitation du gel précurseur. Ces synthèses ont été réalisées à partir d’un gel séché (35°C) et broyé, obtenu par co-précipitation de solutions de chlorure ferrique et de silicate de sodium sans ajout d’acide chlorhydrique (pH de co-précipitation basique). Le gel a ensuite été mis en contact avec de l’eau distillée puis le pH de la suspension est ajusté à 12
avec une solution d’hydroxyde de sodium (1M).
Nagase et al. (1999) ont synthétisé des nontronites-Mg à 100 et 200°C à partir d’un gel Si-Fe-Mg (4/1.7/0.3) mis en contact avec de l’eau distillée et dont le pH a été ajusté avec une solution de NaOH entre 12 et 12.4. Ce gel est obtenu à un pH de 9.9 par co-précipitation de solutions de silicate de sodium, de chlorure ferrique et de chlorure de magnésium. Ces auteurs indiquent que les conditions optimales pour la formation de nontronite-Mg sont : une faible concentration de Fe(III) dans la solution, une température entre 100 et 200°C et un pH très élevé entre 12 et 13 comme précédemment observé par Grauby (1993).
Guillebault (2002) et Delage (2003) ont obtenu des nontronites purement Fe(III) avec des charges tétraédriques liées à des substitutions Si → Fe(III) de 0.74, 0.6 et 0.45 par O10(OH)2 pendant leurs travaux de DEA à l’Université de Poitiers. Ces nontronites ont été synthétisées à 150°C à partir d’un gel précurseur obtenu par co-précipitation de solutions de
chlorure ferreux et de silicate de sodium puis séché et oxydé à 60°C. Le chlorure ferreux a été préféré au chlorure ferrique puisque le Fe(II) est propice à la formation de structures argileuses lors de la co-précipitation du gel précurseur (Harder, 1976; Decarreau & Bonnin, 1986). Ce gel est ensuite dispersé dans de l’eau, puis le pH est ajusté avec une solution de NaOH à 1 M à des valeurs comprises entre 10 et 12.5. Ces travaux ont mis en évidence le rôle du pH sur la charge foliaire de ces nontronites. Néanmoins, la reproductibilité des synthèses
est difficile du fait de l’ajustement manuel du pH avec l’ajout de NaOH. Suite à ces travaux
de DEA, des nontronites de très haute charge (0.75 par O10(OH)2) ont été obtenues entre 75 -
150°C par Decarreau et al. (2008).
Andrieux & Petit (2010) ont synthétisé la solution-solide nontronite-beidellite entre 150 et 200°C. Cette série a été obtenue par traitement hydrothermal d’un gel séché obtenu par co-précipitation de solutions de chlorure ferreux ou ferrique, de chlorure d’aluminium et de silicate de sodium en présence ou non d’acide chlorhydrique. Ces auteurs ont mis en évidence une forte influence de la température et du pH lors de la cristallisation des smectites de la série nontronite-beidellite. Les smectites vers le pôle nontronite requièrent une température de synthèse plus faible et un pH plus élevé que celles obtenues vers le pôle beidellite.
28 Les protocoles expérimentaux utilisés dans cette thèse ont été établis au vu des résultats de la bibliographie et de l’expérience acquise lors des 25 dernières années sur la
synthèse de nontronite à l’Université de Poitiers. Ils utilisent un gel précurseur précipité à
partir de métasilicate de sodium et de chlorure de fer ferreux ou ferrique. Une température de
150°C a été choisie afin d’obtenir la meilleure cristallinité tout en évitant au maximum la cristallisation de l’aegirine (Decarreau et al., 2004, 2008; Andrieux & Petit, 2010).
L’ajustement du pH lors de la synthèse doit, en revanche, être réalisé différemment afin d’obtenir une reproductibilité des synthèses.
29
Chapitre 2 : Protocoles expérimentaux
1. Synthèses hydrothermales
1.1. Synthèses à partir d’un co-précipité séché
Les synthèses ont été réalisées à partir d’un gel précurseur obtenu par co-précipitation entre une solution de métasilicate de sodium (SiNa2O3.5H2O) à 0.2 M et d’une solution de
chlorure ferreux (Fe(II)Cl2.6H2O) à 0.2 M suivant la réaction suivante :
4 SiNa2O3 + 2 Fe(II)Cl2→ Si4Fe(II)2Na4O12 + 4 NaCl (eq. 2)
Le co-précipité obtenu de couleur gris-bleu est récupéré par filtration et légèrement lavé avec
quelques mL d’eau ultra-pure Milli-Q® (18 MΩ.cm) pendant la filtration afin d’enlever l’excédent de chlorure tout en évitant la dissolution du co-précipité. Le pH de la suspension
après la co-précipitation de 12.55 a été mesuré à 25°C. Pendant la filtration, l’oxydation à l’air ambiant du co-précipité provoque un changement de couleur passant du gris-bleu à un vert-bouteille. Le co-précipité est ensuite complètement oxydé pendant un séchage de 48 heures à 60°C. Le co-précipité de couleur brun est enfin broyé et constitue le gel précurseur.
Le traitement hydrothermal du gel précurseur est réalisé dans des autoclaves métalliques à chemisage Téflon® (Parr®, type: 4744). 500 mg de gel précurseur sont mis en
contact avec 30 ml d’une solution de NaOH à une concentration fixe. Dans cette étude, la
gamme de concentrations de la solution de NaOH est de 10-3 M à 1 M. Les solutions de NaOH ont été obtenues par dilution d’une solution Normadose à 1 M de NaOH dans de l’eau
ultra-pure Milli-Q® (18 MΩ.cm). Le traitement hydrothermal est ensuite effectué à 150°C et
pression de vapeur d’eau saturante (5 bars à 150°C) pendant des durées allant de 1 jour à 183
jours.
Une fois le traitement hydrothermal effectué, l’autoclave est refroidi rapidement. Après la mesure du pH final (pHf), la suspension est filtrée (0.1 µm) afin de collecter la phase
solide. Cette phase est ensuite séchée à 50°C puis broyée.
Dans un souci de reproductibilité de la méthode, le même protocole a été réalisé à
partir d’une autre source de Fe(II). Le co-précipité a été obtenu à partir d’une solution de
métasilicate de sodium (SiNa2O3.5H2O) à 0.2 M et d’une solution de sulfate ferreux
(Fe(II)SO4.7H2O) à 0.2 M suivant la réaction suivante :
4 SiNa2O3 + 2 Fe(II)SO4→ Si4Fe(II)2Na4O12 + 2 Na2SO4 (eq. 3)
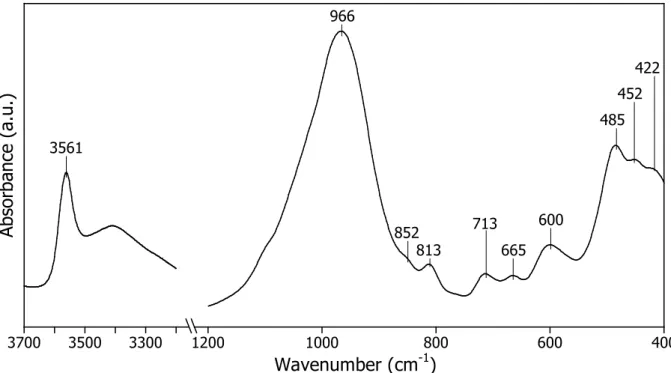
Les spectres infrarouge dans le domaine moyen (MIR) réalisés sur les deux gels (co-précipités à partir de FeCl2 ou FeSO4) montrent rigoureusement les mêmes bandes d’absorption
indiquant une chimie et une structuration similaire (Figure 8a).
Les synthèses hydrothermales nommées NT2 et NT8 ont été réalisées respectivement à partir du gel - FeCl2 et du gel - FeSO4 (Figure 8b). 500 mg de chaque gel a été mis en contact avec 30 ml d’une solution de NaOH à 0.025 M. Les synthèses ont ensuite été réalisées
30 à 150°C pendant 6 jours. Une fois le traitement hydrothermal effectué, l’autoclave est refroidi rapidement. Après la mesure du pH final (pHf), la suspension est filtrée (0.1 µm) afin de
collecter la phase solide. Cette phase est ensuite séchée à 50°C puis broyée.
Figure 8 : (a) Spectres MIR des gels co-précipités à partir de FeCl2 et FeSO4 et (b) spectres MIR des échantillons NT2
synthétisé à partir du gel (FeCl2) et NT8 synthétisé à partir du gel (FeSO4).
Les spectres MIR réalisés sur les échantillons NT2 et NT8 (Figure 8b), montrent
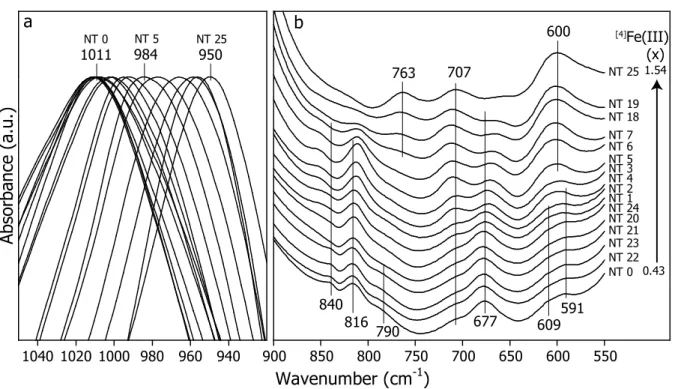
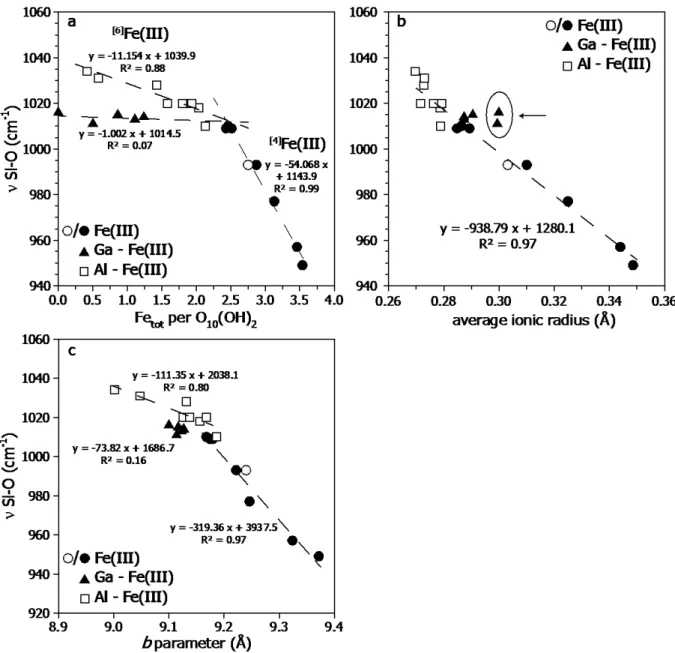
rigoureusement les mêmes bandes d’absorption caractéristiques de la nontronite (Goodman et al., 1976; Decarreau et al., 2008; Petit et al., 2015). La position de la bande à 1001 cm-1
attribuée aux vibrations d’élongation (stretching ( )) du groupement Si-O, dépendant
fortement de la substitution hétérovalente Fe(III) Si(IV) est identique quel que soit
l’échantillon (NT2 ou NT8). Les bandes à 816 et 3560 cm-1 liées respectivement aux vibrations de déformation (bending (δ)) et d’élongation de la liaison OH - Fe3+
2 sont caractéristiques d’un environnement dioctaédrique typique de la nontronite (Goodman et al., 1976; Decarreau et al., 2008; Petit et al., 2015). Ce test montre une très bonne reproductibilité de la méthode afin d’obtenir des nontronites avec une cristallochimie contrôlée.
Le traitement hydrothermal conventionnel (à l’étuve) comme décrit précédemment,
peut être substitué par un traitement hydrothermal assisté par micro-ondes. Ce procédé peut
s’avérer très avantageux pour une application industrielle grâce à sa phase de chauffage rapide et uniforme, sa faible consommation électrique et l’augmentation de la capacité des réacteurs
(volume). Le traitement hydrothermal a été réalisé à 150°C pendant 3 jours avec un four micro-ondes Milestone MycroSynth ayant une puissance de 300 W. 500 mg de gel précurseur séché est mis en contact avec une solution de NaOH à 0.25 M. Le spectre MIR (Figure 9)
montre les bandes d’absorption caractéristiques d’une nontronite de très haute charge
(Chapitre 3, §2). Cet essai montre la possibilité de synthétiser des nontronites à partir d’un traitement hydrothermal assisté par micro-ondes.
Wavenumber (cm-1) A bs or ba nc e (a .u .) 1000 2000 3000 gel (with FeCl2)
gel (with FeSO4)
900 1000 1100 1001 NT2 NT8 1000 2000 3000 3560 816 491 677 997 456 680 3555 a b
31
Figure 9 : Spectre MIR de l’échantillon de nontronite réalisé par synthèse hydrothermale assisté par micro-ondes.
Néanmoins, les résultats obtenus sur les synthèses hydrothermales assistées par micro-ondes de minéraux argileux de type 1:1 et 2:1 en comparaison avec le traitement hydrothermal conventionnel (Travaux associés, §1), ne montrent aucun gain de durée de synthèse par voie micro-ondes afin d’obtenir des produits de synthèses similaires à ceux synthétisés par voie conventionnelle. Au vue de ces résultats et de la pénibilité de l’utilisation du four à micro-ondes (coupures électriques récurrentes), cette méthode n’a pas été utilisée pour la suite de la thèse.
1.2. Synthèses à partir d’un co-précipité hydraté
Un deuxième protocole de synthèse a été développé pendant cette thèse. Les synthèses ont dans ce cas été réalisées à partir d’un gel précurseur obtenu par co-précipitation d’une solution de métasilicate de sodium (SiNa2O3.5H2O) à 0.2 M et une solution de chlorure
ferrique (Fe(III)Cl3.(6H2O) à 0.2 M selon la réaction suivante :
4 SiNa2O3 + 2 Fe(III)Cl3→ Si4Fe(III)2Na2O12 + 6 NaCl (eq. 4)
Les quantités de matière sont calculées pour une masse de gel précurseur de formule théorique Si4Fe(III)2Na2O12 anhydre de 300 mg. La suspension obtenue est filtrée afin de
collecter le co-précipité. La totalité du co-précipité est ensuite directement placé sans séchage dans une autoclave métallique à chemisage Téflon® (Parr®, type: 4744). Afin de contrôler la quantité de matière introduite dans l’autoclave, un gel est fabriqué selon le même protocole pour chaque synthèse. Le gel est ensuite dispersé dans 30 mL d’eau ultra-pure Milli-Q® (18
MΩ.cm) ou dans une solution de NaOH à une concentration fixe. Comme le protocole précédent, les solutions de NaOH ont été obtenues par dilution d’une solution Normadose à 1
M de NaOH dans de l’eau ultra-pure Milli-Q®(18 MΩ.cm). La gamme de concentration de la
solution de NaOH utilisée ici est de 10-3 M à 0.25 M. 3300 3500 3700 Wavenumber (cm-1) 400 600 800 1000 1200 A bs or ba nc e (a .u .) 3561 966 852 813 713 665 600 422 485 452
32 Le traitement hydrothermal est effectué à 150°C et pression de vapeur d’eau saturante (5 bars à 150°C) pendant des durées de 6 et 31 jours. Après traitement, l’autoclave est refroidi rapidement. La suspension est ensuite filtrée (0.1 µm) afin de collecter la phase solide. La phase solide a été soit séchée à 50°C puis broyée, soit remise en suspension dans de l’eau ultra-pure Milli-Q®(18 MΩ.cm).
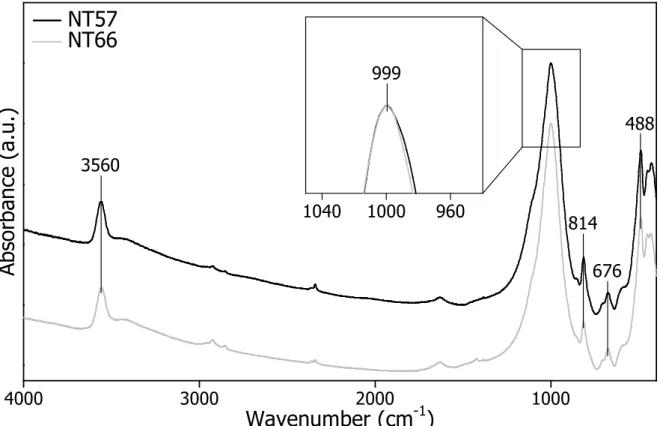
Toujours dans un souci de reproductibilité, la méthode a été testée. Deux synthèses hydrothermales ont été effectuées à partir de deux gels dans des solutions de NaOH à 0.01 M. Les analyses des spectres MIR montrent que les produits finaux sont des nontronites possédant les mêmes caractéristiques cristallochimiques quel que soit l’échantillon (Figure 10).
Figure 10 : Spectres MIR des échantillons NT57 et NT66 synthétisés dans les mêmes conditions.
1.3. Synthèses hydrothermales en milieu réducteur
Les expérimentations ont été menées dans une boîte à gants anaérobique 855-ACB Plas-Labs. Les conditions anaérobiques ont été obtenues en procédant à trois purges avec du diazote (N2) et enfin une dernière purge à l’aide d’un gaz anaérobique composé de 85 % de
N2, de 10 % de dihydrogène (H2), et de 5 % de dioxyde de carbone (CO2). La réduction
maximale de dioxygène (O2) est réalisée par l’introduction, dans la boîte à gants, de pastilles
de palladium qui servent de catalyseurs pour combiner les traces d’O2 avec l’H2 afin de former de la vapeur d’eau et de la chaleur. Pour prévenir des possibles fuites, la boîte à gants
est légèrement mise sous pression.
Pour la synthèse, un gel précurseur a été co-précipité à partir d’une solution de métasilicate de sodium (SiNa2O3.5H2O) à 0.2 M et une solution de chlorure ferreux
(Fe(II)Cl2.6H2O) à 0.2 M suivant la réaction suivante :
![Figure 11 : Powder XRD patterns of the synthesized samples. The [4] Fe(III) content (x) of the synthesized samples [Si 4- 4-x Fe(III) x ] Fe(III) 2 O 10 (OH) 2 Na x increases from the bottom to the top](https://thumb-eu.123doks.com/thumbv2/123doknet/7895999.264418/46.892.111.786.248.626/figure-powder-patterns-synthesized-samples-content-synthesized-increases.webp)

![Figure 19 : Correlation between the [4] Fe(III) content (x) of the synthesized samples [Si 4-x Fe(III) x ] Fe(III) 2 O 10 (OH) 2 M(I) x and the position of the d 06-33](https://thumb-eu.123doks.com/thumbv2/123doknet/7895999.264418/55.892.204.667.562.900/figure-correlation-iii-content-synthesized-samples-iii-position.webp)
![Figure 25 : Position de la bande [6] M 3+ -O-Si en fonction de la teneur en Fe(III) totale de l’échantillon](https://thumb-eu.123doks.com/thumbv2/123doknet/7895999.264418/69.892.220.668.366.692/figure-position-bande-fonction-teneur-iii-totale-échantillon.webp)