Université de Lille
Année Universitaire 2018/2019 Faculté de Pharmacie de Lille
THESE
POUR LE DIPLOME D'ETAT DE DOCTEUR EN PHARMACIE
Soutenue publiquement le jeudi 26 septembre 2019 Par Mme DELERUE Eva
_____________________________
Accès précoce aux médicaments : aspects réglementaires et market access en France et en Europe.
_____________________________
Membres du jury :
Président : Mme PERROY Anne-Catherine Professeur des Universités, Faculté des Sciences Pharmaceutiques et Biologiques de Lille (Université Lille)
Directeur, conseiller de thèse : M. SERGHERAERT Eric Professeur des Universités, Faculté des Sciences Pharmaceutiques et Biologiques de Lille (Université Lille)
Assesseur(s) : Mme LABYT Charlotte Responsable Affaires Réglementaires et Assurance Qualité – Accord Healthcare
Université de Lille
Président : Jean-Christophe CAMART
Premier Vice-président : Damien CUNY
Vice-présidente Formation : Lynne FRANJIÉ
Vice-président Recherche : Lionel MONTAGNE
Vice-président Relations Internationales : François-Olivier SEYS
Directeur Général des Services : Pierre-Marie ROBERT
Directrice Générale des Services Adjointe : Marie-Dominique SAVINA
Faculté de Pharmacie
Doyen : Bertrand DÉCAUDIN
Vice-Doyen et Assesseur à la Recherche : Patricia MELNYK Assesseur aux Relations Internationales : : Philippe CHAVATTE Assesseur à la Vie de la Faculté et aux
Relations avec le Monde Professionnel : Thomas MORGENROTH
Assesseur à la Pédagogie : Benjamin BERTIN
Assesseur à la Scolarité : Christophe BOCHU
Responsable des Services : Cyrille PORTA
Liste des Professeurs des Universités - Praticiens Hospitaliers
Civ. NOM Prénom Laboratoire
Mme ALLORGE Delphine Toxicologie
M. BROUSSEAU Thierry Biochimie
M. DÉCAUDIN Bertrand Pharmacie Galénique
M. DEPREUX Patrick ICPAL
M. DINE Thierry Pharmacie clinique
Mme DUPONT-PRADO Annabelle Hématologie
M. GRESSIER Bernard Pharmacologie
M. LUYCKX Michel Pharmacie clinique
M. ODOU Pascal Pharmacie Galénique
M. STAELS Bart Biologie Cellulaire
Faculté de Pharmacie de Lille
3, rue du Professeur Laguesse - B.P. 83 - 59006 LILLE CEDEX 03.20.96.40.40 - : 03.20.96.43.64
Liste des Professeurs des Universités
Civ. NOM Prénom Laboratoire
M. ALIOUAT El Moukhtar Parasitologie
Mme AZAROUAL Nathalie Physique
M. BERTHELOT Pascal Onco et Neurochimie
M. CAZIN Jean-Louis Pharmacologie – Pharmacie
clinique
M. CHAVATTE Philippe ICPAL
M. COURTECUISSE Régis Sciences végétales et fongiques
M. CUNY Damien Sciences végétales et fongiques
Mme DELBAERE Stéphanie Physique
M. DEPREZ Benoît Lab. de Médicaments et Molécules
Mme DEPREZ Rebecca Lab. de Médicaments et Molécules
M. DUPONT Frédéric Sciences végétales et fongiques
M. DURIEZ Patrick Physiologie
M. FOLIGNE Benoît Bactériologie
M. GARÇON Guillaume Toxicologie
Mme GAYOT Anne Pharmacotechnie Industrielle
M. GOOSSENS Jean François Chimie Analytique
M. HENNEBELLE Thierry Pharmacognosie
M. LEMDANI Mohamed Biomathématiques
Mme LESTAVEL Sophie Biologie Cellulaire
M. LUC Gerald Physiologie
Mme MELNYK Patricia Onco et Neurochimie
M. MILLET Régis ICPAL
Mme MUHR – TAILLEUX Anne Biochimie
Mme
PAUMELLE-LESTRELIN
Réjane Biologie Cellulaire
Mme PERROY Anne Catherine Législation
Mme ROMOND Marie Bénédicte Bactériologie
Mme SAHPAZ Sevser Pharmacognosie
M. SERGHERAERT Eric Législation
Mme SIEPMANN Florence Pharmacotechnie Industrielle
M. SIEPMANN Juergen Pharmacotechnie Industrielle
M. WILLAND Nicolas Lab. de Médicaments et Molécules
Liste des Maîtres de Conférences - Praticiens Hospitaliers
Civ. NOM Prénom Laboratoire
Mme BALDUYCK Malika Biochimie
Mme GARAT Anne Toxicologie
Mme GOFFARD Anne Bactériologie
M. LANNOY Damien Pharmacie Galénique
Mme ODOU Marie Françoise Bactériologie
Liste des Maîtres de Conférences
Civ. NOM Prénom Laboratoire
Mme ALIOUAT Cécile Marie Parasitologie
M. ANTHERIEU Sébastien Toxicologie
Mme AUMERCIER Pierrette Biochimie
Mme BANTUBUNGI Kadiombo Biologie cellulaire
Mme BARTHELEMY Christine Pharmacie Galénique
Mme BEHRA Josette Bactériologie
M BELARBI Karim Pharmacologie
M. BERTHET Jérôme Physique
M. BERTIN Benjamin Immunologie
M. BLANCHEMAIN Nicolas Pharmacotechnie industrielle
M. BOCHU Christophe Physique
M. BORDAGE Simon Pharmacognosie
M. BOSC Damien Lab. de Médicaments et Molécules
M. BRIAND Olivier Biochimie
M. CARNOY Christophe Immunologie
Mme CARON Sandrine Biologie cellulaire
Mme CHABÉ Magali Parasitologie
Mme CHARTON Julie Lab. de Médicaments et Molécules
M CHEVALIER Dany Toxicologie
M. COCHELARD Dominique Biomathématiques
Mme DANEL Cécile Chimie Analytique
Mme DEMANCHE Christine Parasitologie
Mme DEMARQUILLY Catherine Biomathématiques
M. DHIFLI Wajdi Biomathématiques
Mme DUMONT Julie Biologie cellulaire
Mme DUTOUT-AGOURIDAS Laurence Onco et Neurochimie
M. EL BAKALI Jamal Onco et Neurochimie
M. FARCE Amaury ICPAL
Mme FLIPO Marion Lab. de Médicaments et Molécules
Mme FOULON Catherine Chimie Analytique
M. FURMAN Christophe ICPAL
Mme GENAY Stéphanie Pharmacie Galénique
M. GERVOIS Philippe Biochimie
Mme GOOSSENS Laurence ICPAL
Mme GRAVE Béatice Toxicologie
Mme GROSS Barbara Biochimie
M. HAMONIER Julien Biomathématiques
Mme HAMOUDI Chérifa Mounira Pharmacotechnie industrielle Mme HANNOTHIAUX Marie-Hélène Toxicologie
Mme HELLEBOID Audrey Physiologie
M. HERMANN Emmanuel Immunologie
M. KAMBIA Kpakpaga Nicolas Pharmacologie
M. KARROUT Youness Pharmacotechnie Industrielle
Mme LALLOYER Fanny Biochimie
M. LEBEGUE Nicolas Onco et Neurochimie
Mme LECOEUR Marie Chimie Analytique
Mme LEHMANN Hélène Législation
Mme LIPKA Emmanuelle Chimie Analytique
Mme MARTIN Françoise Physiologie
M. MOREAU Pierre Arthur Sciences végétales et fongiques
M. MORGENROTH Thomas Législation
Mme MUSCHERT Susanne Pharmacotechnie industrielle
Mme NIKASINOVIC Lydia Toxicologie
Mme PINÇON Claire Biomathématiques
M. PIVA Frank Biochimie
Mme PLATEL Anne Toxicologie
M. POURCET Benoît Biochimie
M. RAVAUX Pierre Biomathématiques
Mme RAVEZ Séverine Onco et Neurochimie
Mme RIVIERE Céline Pharmacognosie
Mme ROGER Nadine Immunologie
M. ROUMY Vincent Pharmacognosie
Mme SEBTI Yasmine Biochimie
Mme SINGER Elisabeth Bactériologie
Mme STANDAERT Annie Parasitologie
M. TAGZIRT Madjid Hématologie
M. VILLEMAGNE Baptiste Lab. de Médicaments et Molécules
M. WELTI Stéphane Sciences végétales et fongiques
M. YOUS Saïd Onco et Neurochimie
M. ZITOUNI Djamel Biomathématiques
Professeurs Certifiés
Civ. NOM Prénom Laboratoire
M. HUGES Dominique Anglais
Mlle FAUQUANT Soline Anglais
M. OSTYN Gaël Anglais
Professeur Associé - mi-temps
Civ. NOM Prénom Laboratoire
M. DAO PHAN Hai Pascal Lab. Médicaments et Molécules
M. DHANANI Alban Droit et Economie Pharmaceutique
Maîtres de Conférences ASSOCIES - mi-temps
Civ. NOM Prénom Laboratoire
M. BRICOTEAU Didier Biomathématiques
Mme CUCCHI Malgorzata Biomathématiques
M. FRIMAT Bruno Pharmacie Clinique
M. GILLOT François Droit et Economie pharmaceutique
M. MASCAUT Daniel Pharmacie Clinique
M. ZANETTI Sébastien Biomathématiques
M. BRICOTEAU Didier Biomathématiques
AHU
Civ. NOM Prénom Laboratoire
Mme DEMARET Julie Immunologie
Mme HENRY Héloïse Biopharmacie
Faculté de Pharmacie de Lille
3, rue du Professeur Laguesse - B.P. 83 - 59006 LILLE CEDEX Tel. : 03.20.96.40.40 - Télécopie : 03.20.96.43.64
http://pharmacie.univ-lille2.fr
L’Université n’entend donner aucune approbation aux opinions
émises dans les thèses ; celles-ci sont propres à leurs auteurs.
Remerciements
Je souhaite tout particulièrement remercier Madame le Professeur Anne-Catherine PERROY, votre intérêt pour le sujet lors des enseignements que vous nous avez dispensé a été communicatif. Votre disponibilité et votre précieuse aide ont permis d’aboutir à la rédaction de cette thèse dans les meilleures conditions. Un grand merci à Monsieur le Professeur Eric SERGHERAERT, notamment pour vos conseils en amont et votre aide dans le commencement de ce travail de thèse. Merci également à Madame Charlotte LABYT de m’avoir si bien accompagné lors de mes deux derniers stages. Un immense merci d’avoir bien voulu être jury de ce travail de thèse.
Je remercie les pharmaciens qui m’ont accompagné lors de mes études : merci notamment à M. Vigier d’avoir partagé votre passion de la pharmacie en m’ayant ouvert les portes de votre officine. Merci aux pharmaciens du laboratoire Accord Healthcare pour tout ce que j’ai pu apprendre pendant ces 10 mois de stage à vos côtés. Et plus globalement, je souhaite dire merci à toute l’équipe du Cluster France, Belgique et Pays-Bas pour la formation que j’ai reçu à vos côtés.
Je dédie cette thèse :
A ma mère et mon petit frère, vous avez vécu (et parfois subi) ces six années d’études à mes côtés. Je tiens à remercier ma mère pour l’éducation que j’ai reçue et qui a fait de moi celle que je suis aujourd’hui : elle m’a appris à ne jamais perdre confiance en moi mais toujours persévérer afin d’atteindre mes objectifs.
A ma famille, merci pour votre soutien sans faille et vos encouragements qui m’ont permis de trouver ma voie et de m’épanouir dans mes études.
A mes amis, rencontrés au fil du temps. Une pensée particulière à ceux qui m’ont accompagné pendant la PACES, un grand merci pour votre soutien durant cette année.
Table des figures
Figure 1: Cycle de vie du médicament. ... 24
Figure 2 : Délais moyens d’accès au marché en Europe. ... 48
Figure 3 : Délais moyens et médians de la procédure d’évaluation et de fixation du prix de médicaments ayant fait l’objet d’une ATU. ... 49
Figure 4 : Calendrier d’accès au marché du Keytruda® dans le mélanome avancé en 2ème ligne. ... 69
Figure 5 : Calendrier d’accès au marché du Keytruda® dans le cancer bronchique non à petites cellules. ... 71
Table des tableaux
Tableau 1 : Nombre de patients inclus dans une ATUc par année entre 2012 et 2017. ... 29Tableau 2 : Comparaison entre ATU nominative et ATU de cohorte.. ... 34
Tableau 3 : Type d’accès précoce dans certains pays de l’Union Européenne. ... 54
Tableau 4 : Modes d’accès précoce en Allemagne et au Royaume-Uni. ... 55
Tableau 5 : Informations relatives à l’ATU de cohorte du sofosbuvir, au 07/01/2014. ... 61
Liste des abréviations
AMM : Autorisation de Mise sur le Marché
ANSM : Agence Nationale de Sécurité du Médicament et des produits de santé ASMR : Amélioration du Service Medical Rendu
ATU : Autorisation Temporaire d’Utilisation
ATUn/c : Autorisation Temporaire d’Utilisation nominative/de cohorte BfArM : Autorité compétente allemande
BPPV : Bonnes Pratiques de Pharmacovigilance
BRAF : German Federal Institute for drugs and Medical Devices CE : Commission Européenne
CEESP : Commission Evaluation Economique et de Santé Publique CEPS : Comité Economique des Produits de Santé
CHMP : Committee for Medicinal Products Human Use CJUE : Cour de Justice de l’Union Européenne
CNAMTS : Caisse Nationale de l’Assurance Maladie des Travailleurs Salariés CRPV : Centre Régional de Pharmacovigilance
CSP : Code de la Santé Publique CSS : Code de la Sécurité Sociale CT : Commission de la Transparence
DCI : Dénomination Commune Internationale EEA : European Economic Area
EFPIA : European Federation of Pharmaceutical Industries and Associations EMA : European Medicines Agency
EURORDIS : European Rare Diseases Organisation GHS : Groupe Homogène de Séjours
HAS : Haute Autorité de Santé HCC : Hépatite C Chronique INCa : Institut National du Cancer JO : Journal Officiel
LEEM : Les Entreprises du médicament
LFSS : Loi de Financement de la Sécurité Sociale
MHRA : Medicines and Healthcare products Regulatory Agency PARP-1, 2, 3 : poly(ADP-ribose) polymérase
PD1 : Programmed cell Death 1
PDL1/2 : Programmed Death-Ligand 1/2 PRIME : Priority Medicines
PUI : Pharmacie à Usage Intérieur
PUT : Protocole d’Utilisation Thérapeutique et de recueil d’information RTU : Recommandation Temporaire d’Utilisation
RCP : Résumé des Caractéristiques du Produit SFD : Société Française de Dermatologie UCD : Unité Commune de Dispensation
UNCAM : Union Nationale des Caisses d’Assurance Maladie VHC : Virus de l’Hépatite C
Table des matières
REMERCIEMENTS 9
TABLE DES FIGURES 11
TABLE DES TABLEAUX 11
LISTE DES ABREVIATIONS 13
TABLE DES MATIERES 15
INTRODUCTION SUR L’ACCES PRECOCE EN EUROPE 17
1 ANALYSE DU CADRE JURIDIQUE 22
1.1 Le cadre juridique français 22
1.1.1 ATU 25
1.1.1.1 Point de vue réglementaire et sanitaire des ATU 26
1.1.1.1.1 Les conditions générales d’octroi des ATU 26
1.1.1.1.2 Les différents types d’ATU 28
1.1.1.1.2.1 ATU de cohorte 28
1.1.1.1.2.2 ATU nominatives 30
1.1.1.1.3 Les obligations associées aux ATU 34
1.1.1.1.3.1 PUT 34
1.1.1.1.3.2 Obligations de pharmacovigilance 36
1.1.1.2 Aspects financiers des ATU 37
1.1.2 RTU 42
1.1.2.1 Encadrement législatif 42
1.1.2.2 Mise en place d’une RTU 45
1.1.2.3 Prise en charge des RTU 46
1.1.2.4 Obligations pour les industriels 47
1.1.3 Mesures de fixation des prix en faveur des nouveaux médicaments 48
1.1.3.1 Pré-instruction du dossier de fixation du prix 50
1.1.3.2 Procédure d’inscription accélérée 50
1.2 Mécanismes d’accès précoce dans certains pays européens 52
1.2.1 Recommandation de l’EMA sur un usage compassionnel 52
1.2.2 Accès précoces mis en place par une autorité compétente nationale 53
1.2.2.1 Accès précoce en Allemagne et au Royaume-Uni 54
1.2.2.2 Accès précoce en Suède 56
2 EXEMPLES D’APPLICATIONS DU CADRE JURIDIQUE 57
2.1 Accès précoce au Sofosbuvir en France et en Suède 58
2.1.1 ATU en France 59
2.1.2 Compassionate use en Suède 62
2.1.2.1 Rapport du CHMP sur l’usage compassionnel du sofosbuvir 62 2.1.2.2 Conditions d’utilisation, de distribution, population cible et conditions de suivi de la sécurité
2.2 Accès précoce aux extensions d’indications 66 2.2.1 2018 : le cas de Keytruda ® : une RTU pour une extension d’indication 66
2.2.1.1 Délivrance d’une ATU, une AMM puis une RTU 66
2.2.1.2 Délais d’accès au marché en France en comparaison avec d’autres pays Européens dans deux indications 68
2.2.2 2019 : Lynparza ® et Tecentriq® : une ATUc pour une extension d’indication 72 2.2.2.1 ATU de cohorte d’extension d’indication de Lynparza® 72 2.2.2.2 ATU de cohorte d’extension d’indication de Tecentriq® 73 2.2.2.3 Prise en charge du Lynparza® et du Tecentriq® dans le cadre de leur ATUc pour une extension
d’indication 74
2.2.2.3.1 Mise à disposition à titre gratuit dans l’attente des décrets d’application 74 2.2.2.3.2 Décret n° 2019-855 du 20 août 2019 relatif à la prise en charge précoce de certains
produits de santé. 75
3 CONCLUSION 80
3.1 Aspects réglementaires de l’accès précoce aux médicaments en Europe et en France 81 3.2 Aspects market access de l’accès précoce aux médicaments en Europe et en France 82
Introduction sur l’accès précoce en Europe
Au lendemain des premières grandes crises sanitaires telles que celles du Distilbène et de la Thalidomide, il y a eu un souhait d’harmonisation communautaire dans le domaine du médicament. Ainsi, le début de cette harmonisation est marqué par la directive 65/65/CE et renforcé par la directive 2001/83/CE. Le code communautaire porte sur les médicaments à usage humain et dispose qu’avant sa mise sur le marché, tout médicament doit faire l’objet d’une Autorisation de mise sur le marché (AMM), conformément à l’article 6 de la directive 2001/83/CE1 aux
termes duquel : « Aucun médicament ne peut être mis sur le marché d'un État membre sans qu'une autorisation de mise sur le marché n'ait été délivrée par l'autorité compétente de cet État membre, conformément à la présente directive, ou qu'une autorisation n'ait été délivrée conformément aux dispositions du règlement (CE) no 726/2004 »
Cependant, il existe deux grands procédés avec des finalités différentes permettant de déroger à ce principe que nous allons développer en introduction:
- les processus d’accélération des étapes standards de la vie du médicament, - les outils permettant un accès précoce du patient au médicament avant que ce
dernier n’obtienne une AMM dans une indication considérée.
Processus européens d’accélération du développement et de l’obtention de l’AMM d’un médicament
Dans les mécanismes permettant d’accélérer les étapes de la vie du médicament, nous allons brièvement aborder l’adaptive pathway2 et le PRIME (PRIority
MEdicines)3. Le but ici n’est pas de détailler ces outils mais uniquement d’en donner
une vue globale.
D’abord connu sous le nom de « adaptive licensing », l’adaptive pathway a été proposé en 2012 par un groupe d’experts, dans le but de mettre en place une
1 Directive 2001/83/CE du Parlement Européen et du Conseil du 6 novembre 2001 instituant un code
communautaire relatif aux médicaments à usage humain.
2 https://www.ema.europa.eu/en/human-regulatory/research-development/adaptive-pathways 3 https://www.ema.europa.eu/en/human-regulatory/research-development/prime-priority-medicines
nouvelle façon d’accéder à certains médicaments. Puis le concept a changé de nom pour insister sur l’intérêt porté au développement et à l’introduction de nouveaux médicaments.
Ce processus est défini par l’EMA (European Medicines Agency) comme « une approche prospective, planifiée et adaptive pour la mise sur le marché des médicaments »4. Le but est de favoriser les interactions précoces en incitant les
industriels à soumettre leur candidat-médicament dans les premières phases de développement.
Le processus commence par l’obtention d’une AMM précoce et restrictive : qui concerne une population très précise. Pour cette première AMM niche, le laboratoire devra réaliser un développement clinique entier.
Suite à cette première AMM, des phrases de développement itératives auront lieu et seront associées aux données de sécurité et d’efficacité générées par l’utilisation en vie réelle du médicament dans le cadre de son AMM niche.
Ainsi, au final le médicament aura son AMM niche complétée par d’autres indications, donnant une AMM plus complète.
En 2016, l’EMA a publié son compte-rendu de la phase pilote lancée en 2014 et a annoncé que le concept des adaptive pathways allait faire partie intégrante du système réglementaire Européen.
L’EMA a réalisé un atelier sur les adaptive pathways et a publié un rapport sur une réunion en présence des parties prenantes le 8 décembre 2016.5
Dans ce rapport, l’EMA insiste sur le fait que les adaptive pathways ont été initiés pour permettre le développement de molécules de façon à mieux répondre aux besoins des patients. La différence avec les voies standards de mise sur le marché est uniquement le développement, les médicaments continueront d’être autorisés selon les mêmes règles avec notamment les mêmes exigences quant à l’évaluation du rapport bénéfice/risque.
Les adaptives pathways permettent un élargissement des outils mis à disposition pour générer des preuves. En ce sens, les adaptive pathways proposent l’utilisation de données de vraie vie. Ces données ne sont pas là pour remplacer les essais
4 https://www.ema.europa.eu/en/documents/other/pilot-project-adaptive-licensing_en.pdf 5
cliniques mais pour venir les compléter afin de générer plus d’informations dans l’intérêt du patient. Il faut garder à l’esprit que les essais cliniques randomisés auront toujours un rôle central dans l’évaluation des médicaments.
Un autre mécanisme communautaire existe pour obtenir une AMM de façon plus rapide, il s’agit du PRIME, lancé en mars 2016 par l’EMA. Le PRIME a pour but d’apporter aux patients, de façon plus rapide, des médicaments innovants en optimisant et en encourageant le développement de ces traitements. Ainsi, tous les médicaments ne sont pas éligibles à cet outil, ils doivent répondre à un besoin médical non satisfait. En effet, le médicament doit apporter un bénéfice thérapeutique majeur au patient par rapport aux traitements déjà autorisés ou alors, le médicament doit être destiné à des patients en impasse thérapeutique.
Quand le médicament répond à ce critère d’éligibilité et que des données préliminaires sont disponibles et prouvent que le médicament répond à un besoin médical non satisfait, alors l’EMA va apporter son soutien afin d’optimiser le développement de ce produit et l’EMA va accélérer son évaluation afin de permettre aux patients d’avoir le traitement rapidement.
Dans son état des lieux de deux années selon le schéma PRIME6, l’EMA explique
comment elle a soutenu les produits éligibles à ce mécanisme : par exemple, des réunions précoces ont été organisées avec un rapporteur du CHMP (Committee for Medicinal Products for Human Use) afin de préparer un dossier robuste pour la demande d’AMM et une personne de l’EMA dédiée au PRIME a permis de coordonner le soutien règlementaire apporté pendant le PRIME.
Ainsi, l’EMA conclut que le PRIME continue de remplir ses promesses en termes de performance. D’autres analyses du schéma PRIME seront publiées et permettront de dresser des conclusions plus complètes.
Au niveau européen, en dehors des possibilités d’accélérer les étapes de la vie d’un médicament, des mécanismes réglementaires permettent aux patients d’avoir accès précocement à certains médicaments de façon dérogatoire, cela avant l’obtention de l’AMM. C’est précisément le thème de ce travail de thèse.
Outils européens permettant un accès précoce aux médicaments
L’article 5(1) de la directive 2001/83/CE7 donne un cadre légal aux Etats Membres
afin qu’ils puissent fournir des médicaments ne disposant pas d’une AMM pour répondre à des demandes spontanées de médecins pour leurs patients. Cet article dispose que « Un Etat membre peut, conformément à la législation en vigueur et en vue de répondre à des besoins spéciaux, exclure des dispositions de la présente directive les médicaments fournis pour répondre à une commande d’un professionnel de santé agréé et destinées à ses malades particuliers sous sa responsabilité personnelle directe ».
De plus, l’article 83 du règlement européen n° 726/20048 laisse la possibilité aux
Etats Membres de rendre disponible, dans le cadre d’un usage compassionnel, un médicament qui fait l’objet d’une demande de mise sur le marché ou qui est utilisé dans des essais cliniques. L’usage compassionnel est défini comme le fait de mettre à disposition un médicament à un groupe de patients atteint d’une pathologie chronique, grave ou mettant en jeu le pronostic vital et qui ne peut pas être traité de façon satisfaisante par un médicament déjà autorisé.
Ce même article demande aux Etats Membres de notifier l’EMA de leur utilisation de cet usage compassionnel lorsqu’ils y ont recourt. Notons que les laboratoires ne peuvent pas directement demander l’avis du CHMP sur l’usage compassionnel, ce sont les autorités nationales de santé qui doivent faire la demande.9
Afin de faciliter la mise en place de cet article 83 du règlement européen n°726/2004, l’EMA met à disposition une ligne directrice sur l’usage compassionnel des médicaments10.
7 Directive 2001/83/CE du Parlement Européen et du Conseil du 6 novembre 2001 instituant un code
communautaire relatif aux médicaments à usage humain
8 Règlement (CE) du Parlement Européen et du Conseil du 31 mars 2004 établissant des procédures
communautaires pour l’autorisation et la surveillance en ce qui concerne les médicaments à usage humain et à usage vétérinaire, et instituant une Agence européenne des médicaments
9 STAMP (Safe and Timely Access to Medicines for Patients) Commission Expert Group 10 march
2016, European Commission. STAMP 4/22 on compassionate use programmes.
10
Le but de l’usage compassionnel est de faciliter l’accès à de nouvelles thérapies qui sont en cours de développement.11
Cet usage est soumis à des conditions strictes sous lesquelles un médicament peut être mis à disposition des patients, lorsque les critères suivants sont remplis : - Il est destiné au traitement d’une maladie chronique, grave ou engageant le
pronostic vital, sans alternative thérapeutique autorisée dans l’Union européenne, - Il concerne un groupe de patients,
- Le médicament doit être utilisé dans un essai clinique ou être en cours d’approbation pour obtenir une AMM centralisée,
- Le médicament doit faire partie du champ obligatoire de la procédure centralisée.
L’EMA établit des recommandations via le CHMP mais l’usage compassionnel est donc coordonné par chaque état membre mettant en place ses propres règles.
L’usage compassionnel n’a pas pour but de remplacer les essais cliniques qui représentent le seul moyen d’obtenir des données fiables en termes d’efficacité et de sécurité. Même si des données de sécurité sont collectées pendant un usage compassionnel, ce type de programme ne doit donc pas remplacer des essais cliniques à but d’évaluation.
En ce sens, l’usage compassionnel ne doit pas ralentir la mise en place d’un essai clinique ayant pour but de fournir des données sur le rapport bénéfice/risque d’un médicament.12
L’usage compassionnel n’est pas non plus de l’usage hors AMM. En effet, l’utilisation d’un médicament hors-AMM correspond au traitement d’un patient avec un médicament, pour une indication autre que celle pour laquelle le médicament a reçu une AMM. Alors que dans l’usage compassionnel, la spécialité pharmaceutique doit être dans un essai clinique ou en cours d’approbation pour obtenir une AMM.
11 Development support and regulatory tools for early access to medicines, 1 march 2016,
EMA/531801/2015
12 Guideline on compassionate use of medicinal products, pursuant to article 83 of regulation (EC) No
Ainsi, deux types de mécanismes sont à différencier : les mécanismes permettant d’accélérer l’évaluation d’un médicament et des procédés d’accès précoce mettant ainsi le médicament à disposition avant qu’il ne dispose d’une AMM.
Au niveau européen, à travers la deuxième partie de l’introduction, nous venons de montrer qu’il existe des outils permettant un accès précoce, ceci dans le but de mettre en place un cadre harmonisé. Cependant, chaque Etat-Membre est libre de mettre en place nationalement des mécanismes permettant l’accès précoce. En ce sens, nous allons analyser le cadre juridique (1) en détaillant le cadre juridique français avec ses mécanismes nationaux d’accès précoce (1.1) puis nous étudierons les mécanismes d’accès précoce mis en place dans certains pays européens (1.2).
Dans un deuxième temps nous étudierons comment est mis en application le cadre juridique (2) au travers de divers exemples.
1 Analyse du cadre juridique
Nous allons présenter les dispositifs français d’accès précoce mis en place nationalement pour venir compléter le cadre européen (1.1) puis nous ferons un état des lieux des mécanismes d’accès précoce établit dans d’autres pays européens (1.2).
1.1 Le cadre juridique français
L’obligation, posée par le Code communautaire, de disposer d’une AMM pour qu’un médicament puisse être commercialisé, est transposée à l’article L. 5121-8 du CSP (Code de la Santé Publique) qui dispose : « Toute spécialité pharmaceutique ou tout autre médicament fabriqué industriellement ou selon une méthode dans laquelle intervient un processus industriel ainsi que tout générateur, trousse ou précurseur qui ne fait pas l'objet d'une autorisation de mise sur le marché délivrée par l'Union européenne en application du règlement (CE) n° 726/2004 du Parlement européen et du Conseil, du 31 mars 2004 (…) doit faire l'objet, avant sa mise sur le marché ou sa distribution à titre gratuit, d'une autorisation de mise sur le marché
délivrée par l'Agence nationale de sécurité du médicament et des produits de santé. » Cet article instaure la nécessité d’avoir une AMM, qui peut être délivrée par l’agence européenne du médicament ou par une agence nationale, donc l’Agence nationale de sécurité du médicament et des produits de santé (ANSM) en France.
Cependant, la possibilité, ouverte par le droit communautaire, de déroger à l’obligation d’obtenir une AMM est prévue par le droit français.
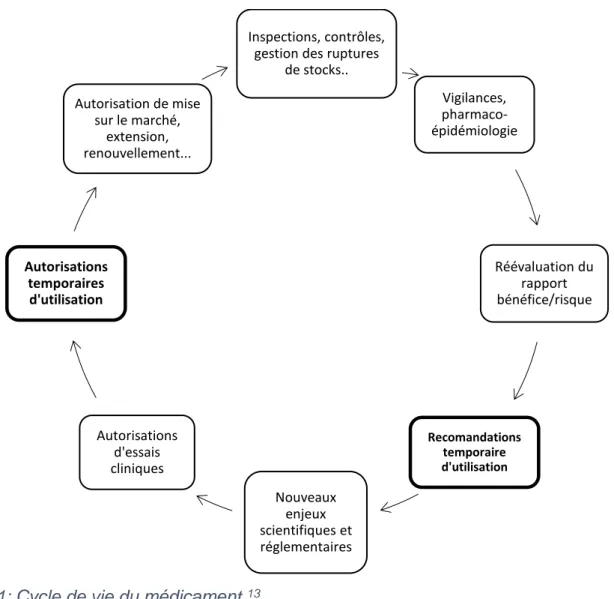
La figure ci-dessous (Figure 1) replace, dans le cycle de vie d’un médicament, en France, les possibilités d’utilisation d’un médicament n’ayant pas encore fait l’objet d’une AMM pour l’indication concernée.
Le cycle de vie d’un médicament débute par une idée aboutissant à des candidats médicaments qui vont faire l’objet d’essais cliniques. Selon les résultats de ces essais cliniques et l’urgence du besoin thérapeutique, le laboratoire ou un médecin hospitalier, le cas échéant, pourra solliciter une Autorisation temporaire d’utilisation (ATU) pour ce médicament afin de permettre un accès précoce dans l’attente de l’obtention d’une AMM. Suite à l’obtention de cette dernière et à la commercialisation du médicament, la pharmacovigilance sera mise en place pour surveiller le médicament pendant la période post AMM. Lors de cette période post AMM, l’ANSM pourra décider, le cas échéant, de mettre en place une RTU (Recommandation Temporaire d’Utilisation) pour le médicament, dans une indication donnée, afin d’encadrer des usages hors AMM.
Figure 1: Cycle de vie du médicament.13
Ainsi, en France, dans le cycle du médicament il existe une option réglementaire permettant un accès précoce aux médicaments, il s’agit de l’ATU.
Parallèlement à cet outil, dans le but de sécuriser l’utilisation des médicaments prescrits dans un cadre hors-AMM, l’ANSM peut décider de mettre en place des RTU. Ce mécanisme n’a pas été créé pour être un outil d’accès précoce mais peut être utilisé en tant que tel en pratique dans certains cas.
Premièrement, nous verrons qu’en France, à titre exceptionnel, par procédure dérogatoire, l’ANSM délivre des ATU pour des médicaments ne possédant pas d’AMM (1.1.1).
13 Le système des ATU en France, Journée de réflexion scientifique 2013. A. Dhanani
Inspections, contrôles, gestion des ruptures
de stocks.. Vigilances, pharmaco-épidémiologie Réévaluation du rapport bénéfice/risque Recomandations temporaire d'utilisation Nouveaux enjeux scientifiques et réglementaires Autorisations d'essais cliniques Autorisations temporaires d'utilisation Autorisation de mise sur le marché, extension, renouvellement...
Dans un second temps, nous allons aborder les RTU, délivrées par l’ANSM pour une nouvelle indication d’un médicament disposant déjà d’une AMM dans une autre indication. (1.1.2). Enfin, nous étudierons dans cette partie les possibles mesures mises en place pour la fixation du prix des nouveaux médicaments (1.1.3).
1.1.1 ATU
En France, pour qu’un patient puisse bénéficier d’un traitement, ce dernier doit posséder une AMM ou faire l’objet d’un essai clinique, comme mentionné précédemment.
Cependant, depuis le décret du 8 juillet 199414, une dérogation à la procédure
d’AMM existe, le principe étant posé par l’article L. 5121-12 du CSP aux termes duquel :
« l'utilisation, à titre exceptionnel, de certains médicaments, dans des indications thérapeutiques précises, destinés à traiter des maladies graves ou rares, en l'absence de traitement approprié, lorsque la mise en œuvre du traitement ne peut pas être différée et que l'une des conditions suivantes est remplie :
1° L'efficacité et la sécurité de ces médicaments sont fortement présumées au vu des résultats d'essais thérapeutiques auxquels il a été procédé en vue d'une demande d'autorisation de mise sur le marché qui a été déposée ou que l'entreprise intéressée s'engage à déposer dans un délai déterminé (…) ;
2° Ces médicaments, (…) sont prescrits, sous la responsabilité d'un médecin, à un patient nommément désigné et ne pouvant participer à une recherche impliquant la personne humaine dès lors qu'ils sont susceptibles de présenter un bénéfice pour lui et que leur efficacité et leur sécurité sont présumées en l'état des connaissances scientifiques. Le médecin prescripteur doit justifier que le patient, son représentant légal ou la personne de confiance (…) a reçu une information adaptée à sa situation sur l'absence d'alternative thérapeutique, les risques courus, les contraintes et le bénéfice susceptible d'être apporté par le médicament. »15
14 Décret n° 94-568 du 8 juillet 1994 relatif aux autorisations temporaires d'utilisation de certains
médicaments à usage humain et modifiant le code de la santé publique.
Nous allons premièrement détailler cet article du CSP au travers de l’analyse des ATU d’un point de vue réglementaire et sanitaire (1.1.1.1) puis ensuite nous allons étudier les aspects financiers des ATU (1.1.1.2).
1.1.1.1 Point de vue réglementaire et sanitaire des ATU
Dans cette partie consacrée au point de vue réglementaire et sanitaire des ATU nous allons décrire les conditions générales d’octroi des ATU (1.1.1.1.1) puis développer les différents types d’ATU (1.1.1.1.2), de cohorte (1.1.1.1.2.1) et nominatives (1.1.1.1.2.2). Finalement nous aborderons les obligations associées aux ATU (1.1.1.1.3).
1.1.1.1.1 Les conditions générales d’octroi des ATU
Un médicament peut être mis à disposition des patients, de façon temporaire et dérogatoire, s’il bénéficie d’une ATU. Cette autorisation peut être délivrée si la spécialité remplie les conditions suivantes, mentionnées dans l’article L. 5121-12 du CSP :
- La spécialité est destinée à traiter, prévenir ou diagnostiquer des maladies graves ou rares,
- Il n’y a pas d’alternative thérapeutique appropriée sur le marché, - La mise en œuvre ne peut pas être différée,
- L’efficacité et la sécurité d’emploi de la spécialité sont présumées en l’état des connaissances scientifiques.
Les groupes de pathologies les plus concernées par des ATU sont les maladies neurologiques, les maladies infectieuses (sida, hépatites virales…), le domaine de la cancéro-hémato et les maladies métaboliques.
Même si la France a été pionnière en termes d’accès précoce aux médicaments innovants, le dispositif Français d’accès précoce se devait d’évoluer : l’article 65 de
la LFSS (Loi de Financement de la Sécurité Sociale) 201916 fait ainsi écho aux
débats associés à l’arrivée des immunothérapies anti cancéreuses présentant des extensions d’indication prometteuses. Comme expliqué dans le rapport du Sénat, les nouveaux anticancéreux « visent à renforcer le système immunitaire du patient en agissant sur des récepteurs présents dans différents organes, et non à cibler les cellules cancéreuses. (…) Ils peuvent donc être efficaces, de manière transversale, sur plusieurs types de cancer différents, là où les chimiothérapies visent généralement un organe en particulier. Ce mode d’action a notamment permis le développement d’indications parallèles ou successives pour les anticorps anti-PD1 et anti-PDL1. Les premières AMM pour ces produits (…) ont ainsi successivement concerné le mélanome de stade IV, le cancer du poumon, les lymphomes de Hodgkin, les cancers de la vessie, du rein, de la tête et du cou »17.
L’article 65 de la LFSS 2019 vise ainsi à renforcer l’accès précoce aux médicaments innovants, en venant élargir les conditions d’octroi des ATU. Jusqu'à lors, il n’était pas possible de demander une ATU pour un médicament ayant déjà une AMM pour une ou plusieurs indications. L’ATU était ainsi réservée à la première indication en développement d’un médicament.
Maintenant, cet article permet l’accès précoce à certains médicaments dans le cadre d’une extension d’indication.
Le mécanisme de l’ATU permet, en général, une mise à disposition de la spécialité pharmaceutique neuf à douze mois avant l’obtention de l’AMM.18
Notons que le délai entre l’ATU de cohorte et l’octroi de l’AMM est de plus en plus bref, certaines ATU de cohorte étant octroyées pour des médicaments à un stade avancé de leur développement clinique montrant une sécurité et une efficacité très fortement présumées au travers de l’avis donné par le CHMP.
16 Article 65 de la loi 2018-1203 du 22 décembre 2018 de financement de la sécurité sociale 17 https://www.senat.fr/rap/r17-569/r17-569.html
1.1.1.1.2 Les différents types d’ATU
Après avoir présenté les conditions d’octroi générales pour les ATU, nous allons voir qu’il en existe deux types, nous allons dans un premier temps étudier les conditions d’octroi spécifiques aux ATU dite de cohorte (1.1.1.1.2.1) puis nous allons aborder celles des ATU dite nominatives (1.1.1.1.2.2).
1.1.1.1.2.1 ATU de cohorte
L’ATU de cohorte est demandée par un laboratoire exploitant qui va faire la démarche pour un groupe de patients répondant à certains critères qui seront définis dans l’ATU de cohorte (pathologie, indication(s) thérapeutique(s)…). Les médicaments entrant dans une ATU de cohorte ont une efficacité ainsi qu’une sécurité fortement présumées.
Pour réaliser une demande d’ATU de cohorte recevable, l’exploitant doit : - Soit faire une demande d’AMM simultanément ;
- Soit s’engager à déposer une demande d’AMM ultérieurement dans un délai fixé.
Le laboratoire adresse sa demande d’ATU de cohorte à l’ANSM via le formulaire de demande prévu par l’arrêté du 6 juillet 2007 (annexe 1).
L’article R. 5121-68 du CSP19 définit ce que doit comporter le dossier de demande
d’ATU de cohorte. Il contient notamment :
- Le formulaire de demande d’ATU de cohorte avec les motifs de la demande et l’engagement à déposer une demande d’AMM dans le délai fixé.
- Le dossier administratif, le cas échéant la copie de la demande d’AMM. Ce dossier contient le projet du résumé des caractéristiques du produit (RCP), de notice d’information des patients et d’étiquetage en langue française, le projet de protocole d’utilisation thérapeutique et de recueil d’information (PUT), des informations sur les recherches biomédicales en cours, des informations sur le médicament s’il est autorisé à l’étranger, toute information sur une utilisation exceptionnelle et précoce à l’étranger, la copie de l’avis scientifique délivré par l’ANSM, l’EMA ou toute autre
autorité compétente le cas échéant, ainsi que le nombre de patients en France estimé être concerné par la demande.
- Le dossier technique relatif à la spécialité pharmaceutique avec toutes les données pharmaceutiques et pharmaco-toxico-cliniques qui sont disponibles. Le dossier doit être dans un format le plus proche possible de celui d’un dossier d’AMM.
L’évaluation des demandes d’ATU de cohorte est réalisée par la Direction Produits de la gamme thérapeutique concernée par la demande. L’évaluation de la demande d’ATU de cohorte est réalisée en prenant en compte la qualité pharmaceutique, la sécurité d’emploi et l’efficacité du médicament reflétée par les documents fournis dans le dossier administratif, ainsi que la présence ou non d’alternative thérapeutique sur le marché français.
Lorsqu’une ATU de cohorte est accordée, elle l’est pour une durée d’un an et pourra être renouvelée ou encore être modifiée, suspendue ou retirée par le directeur général de l’ANSM.
L’ANSM met en place une politique de développement de ATU de cohorte comme le montre son rapport d’activité de 201620 : « L'ANSM développe depuis 2012 une
nouvelle politique dont l'objectif est de privilégier, pour tous les patients en situation d'impasse thérapeutique, un accès équitable et encadré aux traitements innovants, par le développement des ATU de cohorte ».
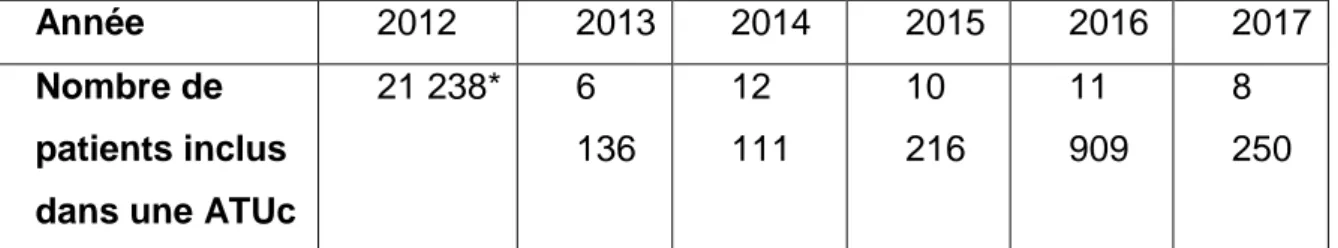
Le tableau ci-dessous montre le nombre de patients inclus dans le cadre d’ATU de cohorte de 2012 à 2017. Ce tableau a été constitué avec les informations du rapport d’activité de l’ANSM de 2016 et de 2017.
Année 2012 2013 2014 2015 2016 2017
Nombre de patients inclus dans une ATUc
21 238* 6 136 12 111 10 216 11 909 8 250
Tableau 1 : Nombre de patients inclus dans une ATUc par année entre 2012 et 2017.21
20 Rapport d’activité 2016 de l’ANSM 21 Synthèse d’activité 2017, ANSM.
* Le nombre de patients inclus dans une ATUc pour l’année 2012 est très élevé du fait de l’ATUc APROKAM, dans laquelle 17 000 patients ont été inclus.
On constate à travers ce tableau, la politique de développement des ATUc menée par l’ANSM.
Nous allons maintenant développer les conditions d’octroi spécifiques aux ATU nominatives (1.1.1.1.2.2.)
1.1.1.1.2.2 ATU nominatives
Les ATU nominatives peuvent permettre un accès précoce à des traitements innovants avant leur commercialisation pour des patients nommément désignés. Contrairement aux ATU de cohorte, les ATU nominatives permettent en effet un accès au cas par cas, ce qui signifie que l’accès pourra alors potentiellement être fait de façon encore plus précoce que dans le cadre des ATU de cohorte. Les ATU nominatives peuvent être délivrées alors que des essais cliniques sont encore en cours et interviennent donc très en amont de la commercialisation d’un médicament.22
Une ATU nominative est demandée par un médecin hospitalier pour un patient nommément désigné, ne pouvant pas être inclus dans un essai clinique. L’ATU nominative est donc accordée pour un patient, un médecin et un médicament. Le bénéfice, la sécurité et l’efficacité du traitement doivent être présumés.23
L’ANSM ne délivre d’ATU nominative uniquement : - S’il y a eu une demande d’ATU de cohorte,
- Ou une demande d’AMM pour le médicament en question (ou si le laboratoire s’engage à faire cette demande),
- Ou s’il y a un essai clinique en cours en France (ou si cet essai est demandé).
Rapport d’activité 2016, ANSM.
22 https://www.senat.fr/rap/r17-569/r17-569.html
Dans un but d’augmentation de la transparence et de modernisation, depuis le 17 septembre 2018, l’ANSM a mis en place de nouvelles modalités de traitement des demandes d’ATU nominatives de médicament.
Les demandes d’ATU nominatives sont maintenant réalisées de façon dématérialisée. En effet, les ATU sont réservées au milieu hospitalier et du fait de l’évolution des technologies, les prescripteurs hospitaliers sont habitués à avoir recours à des demandes dématérialisées. Ce nouveau système permet de simplifier et de sécuriser le traitement et l’analyse des données ainsi que la notification des effets indésirables.
De plus, ces nouvelles modalités de traitement des demandes d’ATU permettent de garantir une meilleure traçabilité des données par rapport à l’ancien système, qu’était le fax.
Dorénavant, les prescripteurs pourront utiliser un guichet unique pour faire les demandes d’ATU nominatives afin de simplifier ces demandes ainsi que les échanges entre l’ANSM et les médecins prescripteurs.
Une application web appelée e-saturne a été mise en place en mars 201924. Cette
plateforme permet une identification précise du prescripteur et des pharmaciens grâce à une connexion sécurisée et un système de signature électronique.
Le formulaire de demandes d’ATU nominatives a quant à lui, été mis à jour. En effet, l’ANSM a mis en place un nouveau formulaire qui intègre les nouvelles dispositions. Depuis le 17 septembre 2018, les médecins prescripteurs doivent utiliser ce formulaire pour soumettre leurs demandes.
De plus, l’ANSM a mis à disposition sur son site internet un référentiel des spécialités délivrées dans le cadre des ATU nominatives. Il recense des fiches produit pour la grande majorité des médicaments actuellement disponibles dans le cadre d’une ATU nominative. Dans ces fiches produit on retrouve notamment les critères d’octroi des ATU. Ces critères d’octroi sont ceux auxquels les médecins prescripteurs font souvent face dans les situations cliniques courantes.
On y trouve aussi d’autres informations sur les médicaments, comme le RCP.
24
https://www.ansm.sante.fr/S-informer/Points-d-information-Points-d-information/Pour-un-acces- rapide-a-l-innovation-therapeutique-toutes-les-demandes-d-ATU-nominatives-peuvent-etre-dematerialisees-via-l-application-e-Saturne-Point-d-information
La nouveauté réside dans le fait que lorsqu’une demande d’ATUn est faite pour un médicament présent dans ce référentiel et que la demande est conforme aux critères d’octroi, alors le traitement de la demande sera fait de façon plus rapide. Une fois la demande d’ATU nominative envoyée, la Direction Produit de la gamme thérapeutique concernée va évaluer, individuellement, chaque demande selon les critères du médicament en prenant en compte différents éléments tels que : - La qualité pharmaceutique ;
- La sécurité d’emploi ;
- L’efficacité dans l’indication revendiquée dans la demande ; - L’absence d’alternative thérapeutique appropriée.
Suite à cette évaluation, l’ANSM peut refuser ou délivrer l’ATU.
Dans le cas de la délivrance de l’ATU, l’autorisation mentionne le nom du médicament bénéficiant d’une ATU, les coordonnés du prescripteur, les initiales du patient inclus dans cette ATU, la durée de l’autorisation, les coordonnées de la pharmacie de l’établissement de santé. L’ATU est communiquée au pharmacien qui en informe le prescripteur.
Dans le cas d’un refus de l’ATU, le refus est communiqué au pharmacien qui en informe le prescripteur. Dans un délai de 2 mois, cette décision de refus peut faire l’objet d’un recours gracieux au Directeur Général de l’ANSM et/ou d’un recours contentieux auprès du tribunal administratif compétent.
Le motif du refus de délivrance d’une ATU nominative peut être :
- L’existence d’une alternative thérapeutique bénéficiant d’une AMM en France et disponible sur le marché ;
- Et/ou l’absence de preuves suffisantes pour présumer de l’efficacité et la sécurité de la spécialité pharmaceutique dans l’indication revendiquée ;
- Et/ou quand l’ATU est demandée dans un objectif d’investigation ;
- Et/ou la possibilité pour le patient d’être inclus dans un essai clinique en cours.
L’ATU nominative est délivrée pour une durée spécifiée dans la décision d’autorisation. Cette durée ne peut dépasser un an. Une demande de renouvellement d’ATU peut être effectuée auprès de l’ANSM en cas de besoin de
prolongation du traitement. Les ATU nominatives peuvent, à tout moment, être modifiées, suspendues ou retirées par le directeur général de l’ANSM.
Par dérogation, une ATU nominative (ATUn dérogatoire) peut être délivrée dans les 3 cas suivants :
- Lorsqu’il existe une très forte probabilité pour le patient de subir des conséquences graves, en l’état des thérapeutiques disponibles,
- Pour un médicament dont la commercialisation est arrêtée : si l’indication demandée est autre que celle autorisée et s’il y a de fortes présomptions d’efficacité et de sécurité pour la spécialité pharmaceutique, dans l’indiction demandée, - Dans le cas où une demande d’ATU de cohorte ou une demande d’autorisation
d’essai clinique a été refusée pour l’indication demandée mais qu’il y a un bénéfice individuel pour le patient pour lequel l’ATU nominative est demandée. Le patient et le prescripteur doivent être au courant des motifs du refus.
Pour résumer, voici les principales différences entre les deux types d’ATU :
ATU de cohorte ATU nominative
Demandeur Un laboratoire Un médecin hospitalier
Efficacité et sécurité d’emploi (art. L 5121-12 CSP) « L'efficacité et la sécurité de ces médicaments sont
fortement présumées au vu des résultats d'essais
thérapeutiques »
« leur efficacité et leur sécurité sont présumées en l'état des connaissances scientifiques. » Population Groupe/sous-groupe de patients traités et surveillés selon les critères du PUT « à un patient nommément désigné et ne pouvant participer à une recherche impliquant la personne humaine »25 25 Article L. 5121-12 du CSP.
Délivrance
Sur demande du titulaire des droits d’exploitation qui a ou va demander une AMM dans un certain délai
Sur demande du médecin prescripteur, et sous sa
responsabilité
Tableau 2 : Comparaison entre ATU nominative et ATU de cohorte..
Lorsque toutes les conditions d’octroi sont remplies, une ATU, de cohorte ou nominative selon les cas, peut être délivrée. Cette dernière sera associée à des obligations qui devront être remplies par les industriels. Nous allons maintenant les développer (1.1.1.1.3).
1.1.1.1.3 Les obligations associées aux ATU
Après avoir développé les conditions d’octroi ainsi que les deux différents types d’ATU, nous allons maintenant aborder les obligations pour les industriels au travers du PUT (1.1.1.1.3.1) et du suivi de pharmacovigilance (1.1.1.1.3.2).
1.1.1.1.3.1 PUT
La mise en place d’une ATU de cohorte est subordonnée à la création d’un protocole d’utilisation thérapeutique et de recueil d’information. Quant aux ATU nominatives, elles, sont aussi subordonnées à la conclusion d’un PUT, sauf dans le cas où il s’agit d’ATU nominative dérogatoire.
Ce protocole est rédigé par l’ANSM en collaboration avec le laboratoire exploitant le médicament. Le PUT détermine :
- Les modalités de suivi des patients inclus dans l’ATU,
- Le recueil des données (efficacité, effets indésirables, conditions réelles d’utilisation),
- Les caractéristiques des patients inclus.
Les objectifs de ce protocole sont d’apporter l’information pertinente aux prescripteurs quant à la spécialité et son utilisation et également de prévoir la surveillance des patients. Le but est aussi de permettre un recueil d’informations
concernant l’utilisation réelle du médicament : les patients traités, l’efficacité du médicament, les effets indésirables constatés suite à cette utilisation. Ces informations sont recueillies pour établir un rapport périodique de synthèse.
Le recueil d’informations est principalement fait pour disposer de données concernant des populations non ou insuffisamment représentées dans les essais cliniques précédant la demande d’AMM.
Ce que comporte un PUT est défini à l’article R. 5121-69 du CSP26, notamment :
- Un rappel de l’autorisation délivrée,
- Les critères d’utilisation du médicament selon le RCP, - Les conditions de prescription et de délivrance,
- Les modalités de recueil des données quant aux patients traités, à l’utilisation réelle du médicament, aux effets indésirables graves ou inattendus,
- Les modalités d’information des patients quant au produit.
Conformément à l’article R. 5121-70 du CSP27, le PUT est communiqué aux
médecins, qui en font la demande et qui peuvent prescrire le médicament, aux pharmaciens des PUI (Pharmacie à Usage Intérieur) susceptibles de délivrer le médicament ainsi qu’aux CRPV (Centres Régionaux de Pharmacovigilance) et centres antipoison. La transmission du PUT est faite par l’exploitant du médicament ou par son mandataire le cas échéant.
Toutes les personnes citées dans cet article du CSP : exploitant ou son mandataire, médecins prescripteurs et pharmaciens des PUI doivent respecter les obligations mentionnées dans le PUT.
Notons que l’article R. 5121-73 prévoit que le directeur général de l’ANSM publie sur le site internet de l’agence les PUT conclus. En effet, les industriels ayant interdiction de faire de la publicité pour un médicament sous ATU, la démarche d’information quant aux ATU est faite par l’ANSM.
26 Article R. 5121-59 du CSP. 27 Article R. 5121-70 du CSP.
Dans le cadre des ATU, en accord avec le PUT, il y a une obligation de suivi. Il s’agit d’un suivi organisé de chaque patient : les médecins remontent les informations au laboratoire. L’exploitant du médicament doit envoyer un projet de rapport de synthèse à l’ANSM pour approbation. Ensuite, une fois approuvé, le rapport de synthèse sera transmis aux médecins ayant prescrit le médicament, aux pharmaciens de PUI l’ayant délivré, aux CRPV ainsi qu’aux centres antipoison. La composition du rapport de synthèse est décrite à l’article R. 5121-73-1 du CSP, comme suit :
- Un résumé de toutes les informations recueillies suite à la mise en place du protocole (conditions d’utilisation du médicament, efficacité et sécurité du médicament …),
- Une analyse de rapport bénéfice risque du médicament.
Lorsque l’ATU ne fait pas l’objet d’un PUT28, c’est-à-dire pour les ATU dérogatoires
prévues au titre du IV de l’article L. 5121-12 du CSP, les modalités de suivi et de recueil d’informations sont mentionnées dans l’ATU. En général, il s’agit d’un suivi de pharmacovigilance classique avec des notifications spontanées des effets indésirables par le médecin au CRPV.
1.1.1.1.3.2 Obligations de pharmacovigilance
Dans les Bonnes Pratiques de Pharmacovigilance (BPPV)29 sont mentionnés les
médicaments sous ATU.
Le laboratoire exploitant un médicament faisant l’objet d’une ATU doit déclarer tout effet indésirable suspecté d’être lié au médicament conformément à l’avis aux demandeurs d’Autorisation Temporaire d’Utilisation30. Ce document prévoit que le
suivi de pharmacovigilance des médicaments sous ATU est similaire à celui des médicaments ayant une AMM. Il y a cependant quelques spécificités. Pour les médicaments bénéficiant d’une ATU avec un PUT, les obligations seront alors mentionnées dans ce PUT. Pour les médicaments sous ATU n’ayant pas de PUT,
28 Article R. 5121-70 du CSP.
29 Bonnes pratiques de pharmacovigilance, février 2018, ANSM.
alors les rapports périodiques actualisés de sécurité seront réalisés conformément au modèle fourni par le directeur général de l’ANSM.
Notons que pour les ATU nominatives, lorsqu’il n’y a pas d’exploitant Français, alors le responsable de la pharmacovigilance pour le médicament sous ATU sera le pharmacien de l’établissement de santé important les médicaments sous ATU.
Après avoir étudié les obligations associées aux ATU, nous allons maintenant aborder les aspects financiers de ces dernières (1.1.1.2.)
1.1.1.2 Aspects financiers des ATU
Les médicaments faisant l’objet d’une ATU sont pris en charge à 100% par l’assurance maladie. Ces médicaments sont fournis aux établissements de santé par le laboratoire exploitant, soit à titre gratuit, soit moyennant une indemnité. Le montant des indemnités des ATU est choisi librement par l’exploitant qui doit, conformément à l’article L. 162-16-5-1 du CSS31 (Code de la Sécurité Sociale),
déclarer au CEPS (Comité Economique des Produits de Santé) :
o Le montant de l’indemnité maximale qu’il peut demander aux établissements, ceci dans le mois suivant l’octroi de l’ATU,
o Son chiffre d’affaire pour les médicaments sous ATU et le nombre d’unités délivrées pendant l’année civile précédente, ceci chaque 15 février.
La prise en charge des ATU est faite en sus du GHS (Groupe Homogène de Séjour) et au fil de l’eau. Afin d’être remboursés, les établissements de santé publics et les établissements de santé privés participant au service public hospitalier déclarent les prescriptions de médicaments sous ATU administrés dans leur centre via un outil grâce aux codes UCD (Unité Commune de Dispensation) tandis que les établissements privés ne participant pas au service public hospitalier se voient remboursés après facturation à la Caisse Nationale de l’Assurance Maladie des Travailleurs Salariés (CNAMTS).
31
https://www.legifrance.gouv.fr/affichCodeArticle.do?cidTexte=LEGITEXT000006073189&idArticle=LE GIARTI000037950860&dateTexte=&categorieLien=id
Il est notable que les ATU tendent à devenir moins nombreuses mais à un prix extrêmement élevé du fait de l’apparition d’innovations de rupture : un changement radical bouleversant les usages et les habitudes.
Selon les chiffres du ministère des solidarités et de la santé, en 2013, les dépenses liées aux ATU représentaient 110 millions d’euros alors qu’elles ont atteint un pic à un milliard d’euros en 2014 et en 2016. Ces deux pics s’expliquent de la façon suivante : en 2014 sont arrivés sur le marché de nouveaux traitements pour l’hépatite C, les antiviraux à action directe, et en 2016 des molécules anti-cancéreuses très onéreuses sont arrivées à l’hôpital. 32
Pour faire face à des ATU de plus en plus onéreuses, les LFSS, à partir de 2017, sont venues durcir les aspects financiers des ATU, au-delà du principe, déjà inscrit dans les textes, d’un remboursement par l’industriel de la différence entre le prix in fine négocié avec le CEPS pour le médicament avec AMM et le prix déclaré de l’ATU.
Depuis la LFSS 2017, l’article L. 162-16-5-1 du CSS fixe une limite aux indemnités remboursées à 10 000€ par patient et par an en prévoyant que « Si, au 31 mars de
chaque année,(…), il apparaît que, pour un médicament dont au moins une des indications est prise en charge au titre d'une autorisation temporaire d'utilisation (…), le montant moyen pris en charge par patient à ce titre pour l'année civile précédente excède 10 000 euros, le laboratoire titulaire des droits d'exploitation de ce médicament, (…) reverse aux organismes mentionnés à l'article L. 213-1 du présent code désignés par le directeur de l'Agence centrale des organismes de sécurité sociale, sous forme de remises, la différence entre le chiffre d'affaires facturé aux établissements de santé et le montant de 10 000 euros multiplié par le nombre de patients traités. Le nombre de patients traités, et en conséquence le montant moyen pris en charge par patient, sont déterminés au prorata de la durée de traitement moyenne sur l'année civile considérée. »
Il est ensuite précisé que, par dérogation, lorsque le chiffre d’affaires hors taxes, pour l’année concernée, du médicament est inférieur à 30 millions d’euros, le remboursement de l’ATU n’est pas plafonné.
De plus, toujours dans l’article L. 162-16-5-1 du CSS, la LFSS 2017 est venue modifier le montant pris en compte dans le calcul de la remise que le laboratoire exploitant doit reverser, remise calculée sur la base de la différence entre le prix déclaré et le prix fixé ultérieurement par négociation avec le CEPS. En effet, depuis cette loi de financement, le laboratoire doit reverser sous forme de remises, la différence entre le montant de l’indemnité déclarée et le prix net de référence de la spécialité, si ce dernier est inférieur à l’indemnité déclarée.
Notons ainsi qu’avant cette loi, le montant des remises à rembourser, en cas de différence entre les montants, était calculé par rapport au prix facial négocié avec le CEPS alors qu’il s’agit maintenant du prix net, qui correspond donc au prix facial moins les remises (comme par exemple les prévisions quant aux volumes de vente). Ceci engendre des coûts plus importants à rembourser, du point de vue des industriels et vient donc durcir le cadre des ATU.
Cette même loi prévoit également désormais un remboursement à la charge du laboratoire (versus un prix de référence fixé par le CEPS), même en l’absence de prix ultérieurement négocié avec le CEPS, à savoir dans les hypothèses de non inscription du médicament sur des listes de remboursement ou en cas d’inscription sur la seule liste des spécialités agréées à l’usage des collectivités.
Réglementairement, l’ATU prend fin lorsque le médicament obtient son AMM (plus précisément 1 à 3 mois après l’AMM, le temps que soit mis à disposition les packs commerciaux).
Toutefois, une fois l’AMM obtenue, le médicament ne peut pas encore être commercialisé. En effet, la phase d’évaluation de la demande de remboursement, le cas échéant, et la négociation du prix sont les étapes restantes précédant l’accès effectif du médicament au marché. . Il est intéressant de noter qu’en France, selon le LEEM (Les Entreprises du médicament), il s’écoule plus de 500 jours entre l’obtention de l’AMM et la publication du prix du médicament au JO (Journal Officiel)33.
Ainsi, mettre fin au mécanisme de l’ATU lorsque le médicament obtient son AMM empêcherait les patients de pouvoir avoir accès à leur traitement et ne permettrait pas d’avoir une prise en charge le temps de la phase d’évaluation par la Commission de la Transparence (CT) de la HAS (Haute Autorité de Santé) et de négociation du prix avec le CEPS. De ce fait, une phase dérogatoire dite de « post ATU » vient en relais de l’ATU pendant cette période afin de permettre une continuité d’accès aux traitements pour les patients ayant bénéficié d’un médicament sous ATU et même, l’inclusion de nouveaux patients au sein des cohortes d’ATU préalablement autorisées.
Dans un premier temps, la loi du 29 décembre 201134, faisant suite à l’affaire
Mediator®, a mis en place un dispositif expérimental permettant la délivrance et la prise en charge des médicaments ayant bénéficié d’une ATU, avant qu’ils n’aient obtenu un remboursement.
Ensuite, la LFSS de 2014 a permis la mise en place définitive de ce dispositif suite à quelques ajustements.
L’article L 162-16-5-2 du CSS, consacré à cette période post-ATU (dont le bénéfice n’est ouvert que si l’industriel a fait une demande de remboursement de son médicament après l’obtention de son AMM), dispose aujourd’hui qu’un médicament ayant bénéficié d’une ATU ayant été prise en charge, « peut, à compter de la date à laquelle l'autorisation temporaire d'utilisation cesse de produire ses effets, être acheté, fourni, pris en charge et utilisé au profit des patients par les collectivités publiques pour l'indication ayant fait l'objet de l'autorisation temporaire d'utilisation dès lors que cette indication est mentionnée dans une autorisation de mise sur le marché délivrée pour ce médicament. »
Il est ensuite précisé que la prise en charge sera décidée à la condition que l’indication n’ait pas fait l’objet d’une évaluation défavorable lors de la demande d’AMM.
Cette prise en charge est effective indication par indication, jusqu’à ce qu’un des évènements suivants survienne :
34 Loi n° 2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé