Mutant protein spread in Huntington's disease and its
implications for other neurodegenerative disorders of
the CNS
Thèse
Alexander Maxan
Doctorat en neurobiologie
Philosophiæ doctor (Ph. D.)
Québec, Canada
Mutant protein spread in Huntington’s disease and its
implications for other neurodegenerative disorders of
the CNS
Thèse
Alexander Maxan
Sous la direction de :
Résumé
La maladie de Huntington (MH) est une maladie génétique dégénérative du système nerveux central (SNC), laquelle se caractérise par une expansion du triplet nucléotidique CAG dans l'exon 1 du gène huntingtine. Cette expansion entraîne la production d’une protéine huntingtine mutée, la mHTT. Cliniquement, la pathologie se manifeste par une variété de symptômes, notamment des problèmes psychiatriques et cognitifs ainsi que des déficits moteurs. Au niveau cellulaire, une atrophie sévère du striatum résulte d'une perte neuronale majeure, laquelle se produit initialement dans le noyau caudé et implique ensuite le putamen. La nature génétique de la MH permet un diagnostic précoce, avant même l’apparition des premiers symptômes. Malgré cela, aucune thérapie neuroprotectrice ou modifiant la maladie ne s’est révélée efficace. Plusieurs essais cliniques de transplantations cellulaires ont été intentés, mais aucun soulagement symptomatique significatif n'a été signalé à ce jour. D’autre part, l'évaluation post-mortem du cerveau de ces patients nous a permis d'identifier des agrégats de mHTT dans le tissu greffé, suggérant que la protéine pourrait être capable de se propager des cellules hôtes aux cellules – pourtant génétiquement saines – du transplant. Nous avons alors émis l'hypothèse que le produit génique anormal, la mHTT, pourrait se propager de cellule à cellule après son introduction dans le cerveau et absorption neuronale. Cette hypothèse a déjà été étayée par de nombreuses publications démontrant les conséquences de la propagation de protéines pathologiques, telles que l’amyloïde β, tau et α-synucléine (α-syn) dans d'autres troubles neurodégénératifs. Dans le cas de la MH, la recherche a été en mesure d'observer ce phénomène in vitro. Au cours de mon doctorat, nous avons entrepris pour la première fois d'évaluer les effets de la propagation de la mHTT in vivo. J’ai tout d’abord pu réaliser l’évaluation post-mortem d'un cerveau de patient atteint de la MH. Je me suis ensuite intéressé à l’étude des conséquences de l'introduction de différents intermédiaires agrégés de la protéine dans le cerveau de différents modèles animaux de la MH (souris et primates non humains). Ces différents protocoles m’ont ainsi permis d'examiner 1- les possibilités d’induire des phénotypes de type MH chez des animaux génétiquement sains, et 2- la capacité à accélérer l’apparition du phénotype chez des modèles transgéniques de la maladie. Pour cela, nous avons administré différentes sources de mHTT – provenant notamment de patients MH, des fibrilles de mHTT exogènes ou encore via l’utilisation d’un virus adéno-associé (AAV) de mHTT – à des souris et des primates non humains. Nos résultats suggèrent fortement que la mHTT peut être libérée dans l’environnement cellulaire. Par la suite, la protéine mutée peut être assimilée par les cellules voisines. Dans le développement de futures thérapies, cette fenêtre de temps entre la libération de la mHTT et sa réabsorption peut se révéler particulièrement opportune pour cibler la protéine mutée. En effet, en ciblant la mHTT extracellulaire, ceci permettrait de réduire la propagation de la protéine et les effets sous-jacents de cette propagation.
Abstract
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder of the central nervous system (CNS) that is characterized by a CAG expansion in exon 1 of the huntingtin gene. This expansion results in the production of mutant huntingtin (mHTT). Clinically, the disease presents a variety of symptoms including psychiatric and cognitive problems in addition to motor disabilities. Neuropathologically, severe atrophy of the striatum results from neuronal loss, which initially occurs in the caudate nucleus and subsequently involves the putamen. The genetic nature of HD allows it to e diagnosed before any signs are observable. Despite this, no effective neuroprotective or disease-modifying therapies have emerged as efficient treatments. My PhD thesis included post-mortem evaluation of an HD patient, but largely focused on introducing different aggregate intermediates of the protein into the brains of mice and non-human primates in order to examine if we can create HD-like phenotypes in wild-type (WT) animals, or if we could accelerate phenotype appearance in transgenic models of the disease. Finally, we hypothesized that the abnormal gene product, mHTT could spread from cell-to-cell following introduction to the brain and neuronal uptake. This has previously been supported by numerous publications demonstrating the consequences of abnormal protein propagation, such as amyloid β, tau and α-synuclein (α-syn) in other neurodegenerative disorders. Research has also been able to examine this phenomenon in vitro, however we set out for the first time to examine the effects of purified mHTT derived from patients, exogenous mHTT fibrils, and an adeno-associated vector delivery of mHTT on the brains of mice and non-human primates. Our results strongly suggest that mHTT can be released and taken up by cells and that this window of time may be opportune for targeting the protein in future therapies. While many current treatments undergoing testing involve lowering the amount of mHTT produced by the gene, these forms of therapies would require treatment extremely early in life as by adulthood, mHTT is already plentiful in the brain. Others aim to target intracellular mHTT, however this fails to acknowledge the importance of extracellular forms of the protein. Thus, the use of antibodies is an interesting avenue to explore. Indeed, these, by targeting the extracellular mHTT, we could reduce the spread of the protein and the underlying effects of this spread.
Table of contents
Résumé ... ii Abstract ... iii Table of contents ... iv List of figures ... ix List of tables ... xiList of abbreviations ... xii
Acknowledgements ... xv Avant-propos ... xvi Introduction... 1 1. Huntington’s disease ... 1 2. Symptomatology ... 1 3. Neuropathology ... 2
4. Proteins in neurodegenerative disorders ... 4
5. The normal form of the HTT protein ... 4
6. The mutated form ... 5
7. Gain vs. loss of function ... 7
8. Animal models ... 8
9. Transcellular propagation ... 10
10. Transynaptic propagation ... 13
11. Hematogenous transport of mHTT through the BBB ... 16
12. Treating HD ... 17
13. Current pharmacological treatments ... 19
14. New avenues of treatment ... 19
14.1. Deep brain stimulation ... 19
14.1. Cell transplantation ... 19
14.1. Antisense oligonucleiotides ... 20
14.1. PRECISION-HD ... 21
15. Hypothesis and objectives ... 22
Chapiter I: Outcome of cell suspension allografts in a patient with Huntington’s disease... 23
1.1 Résumé ... 25
1.4 Materials and methods ... 27
1.4.1 Tissue preparation and neurosurgical procedures ... 27
1.4.2 Post-mortem histological evaluation ... 27
1.4.3 Immunohistochemistry ... 28
1.4.4 Image acquisition and quantification... 28
1.4.5 Statistical analysis ... 29
1.5 Results ... 29
1.5.1 Clinical course ... 29
1.5.2 Post-mortem graft evaluation ... 29
1.5.2.1 Graft location and cytoarchitecture ... 29
1.5.2.2 General graft health ... 29
1.5.2.3 The inflammatory-immune response to cell suspension grafts ... 30
1.5.2.4 Graft innervation and presence of HD-related pathology... 30
1.6 Discussion ... 30
1.7 Funding and acknowledgements ... 31
1.8 Contributions ... 32
1.9 Figures ... 33
Chapter II: Demonstration of prion-like properties of mutant hungtingtin fibrils in both in vitro and in vivo paradigms ... 36
2.1 Résumé ... 37
2.2 Abstract ... 38
2.3 Introduction ... 40
2.4 Materials and methods ... 41
2.4.1 Recombinant HTTExon1Q25 and Q48 fibrils ... 41
2.4.2 Cell lines and culture conditions ... 41
2.4.3 Incubation with fibrils ... 42
2.4.4 Immunofluoresence for in vitro experiments ... 43
2.4.5 Filter retardation assay ... 43
2.4.6 In vitro quantification... 44
2.4.7 Animals ... 44
2.4.8 Intracerebral injections of fibrils ... 45
2.4.9 Intraventricular injections of fibrils ... 45
2.4.11 Behavioral assesment ... 46
2.4.12 Post mortem analyses ... 48
2.4.12.1 Tissue processing ... 48
2.4.12.2 Preparation of plasma free of platelets ... 48
2.4.12.3 Immunofluorescence for in vivo experiments... 48
2.4.13 Enzyme-linked immunosorbent assay ... 49
2.4.14 Image acquisition and preparation... 49
2.4.15 In vivo quantification ... 49
2.4.16 Statistical analysis ... 50
2.5 Results ... 51
2.5.1 Uptaken mHTT Exon1 fibrils of human origin are toxic to multiple cell lines ... 51
2.5.2 Fibrils can recruit WT HTT into aggregates... 52
2.5.3 Intracerebral injection of HTTExon1Q48 fibrils induces cognitive deficits and anxiety-like behavior in WT mice ... 53
2.5.4 Changes in the staining patterns of endogenous HTT in adult WT mice ... 54
2.5.5 Injection of HTTExon1Q48 fibrils precipitates disease in the R6/2 mouse model of HD ... 54
2.5.6 Exogenous HTTExon1Q48 fibrils colocalize with endogenous mHTT in R6/2 mice ... 56
2.5.7 Peripheral injection of mHTTExon1 fibrils initiates an immune response ... 58
2.6 Discussion ... 58
2.7 Funding and acknowledgements ... 61
2.8 Contributions ... 62
2.9 Figures ... 63
Chapter III: Evidence for the spread of human-derived mutant huntingtin protein in mice and non-human primates ... 76
3.1 Résumé ... 77
3.2 Abstract ... 78
3.3 Introduction ... 80
3.4 Materials and methods ... 81
3.4.1 Preparation of human mHTT homogenates for injection into WT mice and non-human primates ... 81
3.4.2 Preparation of human mHTT homogenates for injection into BACHD mice ... 81
3.4.3 Confirmations of homogenate protein content by western blot or dot blot ... 82
3.4.4 Animal husbandry... 83
3.4.6 Behavioral assessment in mice ... 84
3.4.7 Behavioral assessment in non-human primates ... 85
3.4.8 Post mortem analysis ... 85
3.4.9 Image acquisition ... 86
3.4.10 Statistical analysis ... 86
3.5 Results ... 87
3.5.1 Intracerebral injection of human mHTT homogenates does not induce bevhavioral abnormalities in adult WT mice ... 87
3.5.2 mHTT derived from HD brain homogenates are uptaken by neurons and can propagate in the brain of WT mice up to 3 months post-injection ... 88
3.5.3 Intracerebral injection of human mHTT homogenates induces behavioral impairments in adult BACHD mice ... 88
3.5.4 mHTT-derived from HD brain homogenates are present in the brains of BACHD mice 12 months post-injection ... 89
3.5.5 Multiple intracerebral injections of human mHTT homogenates do not induce behavioral nor pathological changes in adult WT non-human primates ... 90
3.5.6 mHTT-derived from HD brain homogenates are present in neurons of non-human primates 8 months post-injection ... 90
3.6 Discussion ... 90
3.7 Funding and acknowledgements ... 93
3.8 Contributions ... 94
3.9 Figures ... 95
Chapter IV: Use of adeno-associated virus-mediated delivery of mutant huntingtin to study the spreading capacity of the protein in mice and non-human primates ... 98
4.1 Résumé ... 99
4.2 Abstract ... 100
4.3 Introduction ... 102
4.4 Materials and methods ... 103
4.4.1 Vector constructs and validation ... 103
4.4.2 In vitro experiments ... 104
4.4.3 In vivo experiments ... 106
4.4.3 Behavioral assessment in mice ... 108
4.4.5 Behavioral assessment in non-human primates ... 108
4.4.6 Post mortem analysis ... 109
4.4.8 Quantification of confocal images... 111
4.4.9 Statistical analysis ... 112
4.5 Results ... 113
4.5.1 Viral transduction with AAV2/6-103Q results in expression and aggregation of mHTT... 113
4.5.2 mHTT aggregates are taken up and released by multiple cell types ... 113
4.5.3 Single intracerebral injection of AAV2/6-103Q induces motor deficits in WT mice ... 113
4.5.4 GFP aggregates are observable up to 14 months post-injection ... 114
4.5.5 Striatal injection of AAV2/6-103Q induces motor deficits in non-human primates ... 114
4.5.6 AAV2/6-Exon1-103Q aggregates are taken up by neurons following viral transduction in non-human primates ... 115
4.5.7 AAV2/6-Exon1-103Q injection decreases endogenous HTT levels following viral transduction in non-human primates ... 116
4.6 Discussion ... 117
4.7 Funding and acknowledgements ... 120
4.8 Contributions ... 120 4.9 Figures ... 121 Conclusion ... 129 1 Observed results... 129 1.1 Objective 1 ... 129 1.2 Objective 2 ... 134 1.3 Objective 3 ... 135 1.4 Objective 4 ... 136
2 Technical considerations and limits ... 138
3 Perspectives ... 141
4 Conclusions ... 143
List of figures
Introduction
Figure 1. The genetics of HD………….………...……..…………...1
Figure 2. Progressive degeneration of the striatum in HD………..…...…….3
Figure 3. The role of protein aggregation in neurodegeneration………..…...….….6
Figure 4. Putative mechanisms of mHTT spreading and seeding capacities………..….………14
Chapter I Figure 1-1. Graft location, cytoarchitecture and grafted cell survival ………...…..33
Figure 1-2. Inflammatory response to cell suspension grafts ………..……….…....….34
Figure 1-3. Graft innervation, presence of HD-related pathology and T-cell response to cell suspension grafts ………..…….………...35
Chapter II Figure 2-1. Uptake of fibrillar HTTExon1Q25 and Q48 by different cell types .……….…...63
Figure 2-2. Seeding of endogenous HTT by exogenous HTtExon1Q48 fibrils .………....….64
Figure 2-3. Manifestation of behavioral impairments in adult WT mice injected with HTTExon1Q48 fibrils………...…..65
Figure 2-4. Post-mortem identification of HTTExon1Q25 and Q48 fibrils in adult WT mice .………..……66
Figure 2-5. Precipitation of behavioral phenotype in R6/2 mice following injection of HTTExon1Q48 fibrils ….67 Figure 2-6. Colocalization of HTTExon1Q48 fibrils and endogenous mHTT in R6/2 mice ……….……...68
Figure 2-7. Post-mortem identification of HTTExon1Q25 and Q48 fibrils in adult WT mice .………..…..…...69
Figure 2-8. Precipitation of behavioral phenotype in R6/2 mice following injection of HTTExon1Q48 fibrils ….70 Figure 2-9. Colocalization of HTTExon1Q48 fibrils and endogenous mHTT in R6/2 mice ………..…….71
Figure 2-10. Post-mortem identification of HTTExon1Q25 and Q48 fibrils in adult WT mice .………....…72
Figure 2-11. Precipitation of behavioral phenotype in R6/2 mice following injection of HTTExon1Q48 fibrils…73 Figure 2-12. Colocalization of HTTExon1Q48 fibrils and endogenous mHTT in R6/2 mice ……….74
Figure 2-13. Colocalization of HTTExon1Q48 fibrils and endogenous mHTT in R6/2 mice ……….…...75
Chapter III Figure 3-1. Cortical injection of human mHTT homogenates in WT mice ………..………...95
Figure 3-2. Cortical injection of human mHTT homogenates in BACHD mice ………..……….…96
Chapter IV
Figure 4-1. AAV constructs and validation ………..…….………..121 Figure 4-2. Detection of aggregate spreading by two-photon intravital imaging ……….………..…...122 Figure 4-3. Behavioral changes in adult WT mice receiving a single cortical AAV2/6-Exon1-103Q injection ………...……….123 Figure 4-4. Reduction in endogenous mouse HTT following a single AAV-103Q-GFP
injection………..……...124 Figure 4-5. Behavioral changes in adult WT non-human primates receiving multiple bilateral striatal AAV2/6-HTTExon1-103Q injections ………..………...125 Figure 4-6. Reduction in endogenous primate HTT following striatal injection of AAV-HTTExon1-103Q-GFP injection ……….………...126
List of tables
Introduction
Table 1. Animal models of HD………...….9 Table 2. Current treatments for HD……….18
Chapter IV
Table 4-1. Summary of vector-mediated expression of mHTT……….…127
Conclusion
List of abbreviations
3-NP: 3-nitropropionic acid -syn: alpha-synuclein A: amyloid beta AC: nucleus accumbens AChE: acethylcholinesterase AD: Alzheimer’s disease AP: antero-posterior
ALS: Amyotrophic Lateral Sclerosis ASOs: antisense oligonucleotides ATP: adenosine triphosphate
BACHD: bacterial artificial chromosome Huntington’s Disease BBB: blood brain barrier
BiFC: biomolecular fluorescence complementation BSA: bovine serum albumin
CAG: cytosine-adenine-guanine
cAMP: cyclic adenosine monophosphate
CAPIT: core assessment program for intracerebral transplantation CB: calbindin
CD4: cluster of differentiation 4 CD8: cluster of differentiation 8 ChAT: choline acetyltransferase CN: caudate nucleus
CNS: central nervous system CPN: cortical pyramidal neurons CR: calretinin
CSF: cerebrospinal fluid CTX M1: primary motor cortex D1: dopamine receptor 1 D2: dopamine receptor 2 DAB: 3,3'-Diaminobenzidine DAPI: 4',6-diamidino-2-phenylindole
DARPP-32: dopamine- and cAMP-regulated phosphoprotein DMEM: Dulbecco’s modified eagle’s media
DPBS: Dulbecco’s phosphate buffered saline DNA: deoxyribonucleic acid
DTT: dithiothreitol DV: dorso-ventral
EAE: experimental autoimmune encephalitis ENK: enkephalin
FDA: Food and Drug Administration
FLVE: far lateral portion of the lateral ventricular eminence FTIR: Fourier transformed infrared spectroscopy
GABA: -aminobutyric acid GFP: green fluorescent protein GFAP: glial fibrillary acidic protein GP: globus pallidus
GPe: external capsule of the globus pallidus GPi: internal capsule of the globus pallidus
HD: Huntington’s disease HPC: hippocampus HPT: hypothalamus HTT: huntingtin
HSP: heat-shock protein
Iba1: ionized calcium binding adaptor molecule 1 IC: internal capsule
Ig: immunoglobulin
iPSC: induced pluripotent stem cell KA: kainic acid
KCl: potassium chloride LB: Lewy body
LGE: lateral ganglionic eminence LV: lateral ventricle
MA: malonic acid
MAP2: microtubule-associated protein 2 MGE: medial ganglionic eminence mHTT: mutant huntingtin
MIG-HD: multicentric intracerebral gtrafting trial in Huntington’s disease miRNA: micro RNA
MgCl2:magnesium chloride
ML: medio-lateral
MRI: magnetic resonance imaging MSNs: medium spiny neurons
NAPDH-d: nicotinamide adenine dinucleotide phosphate-diaphorase NeuN: neuronal nuclei
NHS: N-hydroxysulfosuccinimide NK: natural killer
NMDA: N-methyl-D-aspartic acid NP-zone: non patchy zone PBS: phosphate buffered saline PD: Parkinson’s disease
PET: positron emission tomography PFC: prefrontal cortex
PolyP: polyproline PolyQ: polyglutamine
PREDICT-HD: The Neurobiological Predictors of Huntington’s disease PV: parvalbumin
P-zone: Patchy zone QA: quinolinic acid
RAC-PET: [11C]raclopride positron emission tomography
RNAi: RNA interference SDS: sodium dodecyl sulfate shRNA: short hairpin RNA siRNA: small interfering RNA SNP: single nucleotide polymorphism SOD1: superoxide dismutase 1 TDP-43: TAR DNA-binding protein 43 TFA: trifluoroacetic acid
Tg: transgenic
TNT: tunnelling nanotubes TP: transplantation
UHDRS: Unified Huntington’s disease Rating Scale WGE: whole ganglionic eminence
WT: wild-type
Acknowledgements
I would like to express my sincere thanks to my research supervisor, Dr. Francesca Cicchetti for giving me the opportunity to pursue my education in neuroscience. Her work-ethic was motivational. The time she devoted to helping improve my writing skills, to help me develop a more critical thought process, staying late at night to help with analysis and interpretation, all of it was invaluable and helped me develop immensely. I would like to thank her for giving me the chance to work on such a novel and ground-breaking project, it opened my eyes to techniques and skills I did not previously know existed.
Furthermore, I would like to thank the members of my doctoral committee, Dr. Martin Lévesque, Dr. Ayman El Ali and Dr. Janelle Drouin-Ouellet for accepting to revise my thesis. I am more than honored to have each and every one of you on my thesis defense committee and the value of your revisions was incalculable.
I would like to acknowledge the scholarships that supported me during my graduate studies. Firstly, I would like to thank the FRQS for providing me with a doctoral training award. I would also like to thank the Huntington Study Group for their New Member Award which allowed me to travel internationally to conferences and present my research in front of the most esteemed peers in the field. Lastly, I would like to thank the support I received during my Master’s as well, from the O’Brien Foundation for their fellowship award, and also to Université Laval, for their Bourse de recrutement à la maîtrise fonds Wilbrod-Bhérer.
I would like to express my great appreciation to Martine, Linda and Melanie for their assistance and for guiding me through all aspects of my doctoral studies. Additionally, I would like to thank Giacomo and Alberto for their friendship. You offered companionship and advice through all aspects of life that I can not thank you enough for. Finally, a special thank you to every other member of the Cicchetti team throughout my time here. Your help, suggestions and friendships will forever be valued.
Lastly, I would like to thank my parents, Dwight and Doreen. Your support was never-ending. I could not have done this without you.
Avant-propos
The core of this thesis is composed of 4 main chapters all published or submitted to peer-review journals. Chapters II, III, IV and V consist of the integral versions of 4 articles published between 2018 and 2020. Maxan et al. was published in Annals of Neurology and reported on a post-mortem analysis of a Huntington’s disease patient having received fetal striatal cell-suspension allografts. Masnata et al. was published in Acta
Neuropathologica and investigated the effects of exogenous human fibrillar mHTT (Q48) and HTT (Q25)
N-terminal fragments in three cellular models and three distinct animal paradigms. Gosset et al., is currently under review in Neurobiology of disease and provides evidence for the spread of human-derived mHTT in mice and non-human primates. Maxan et al., is also currently under review in Neurobiology of disease and describes the use of adeno-associated virus-mediated delivery of mutant huntingtin to study the spreading capacity of the protein in mice and non-human primates.
I would also like to point out that in the course of my PhD, I had the opportunity to work on additional projects not directly in line with my main research theme. In particular, I have been involved in a project researching the presence of tau pathology within foetal neural allografts in patients with Huntington’s and Parkinson’s disease. These results were published in Brain (Cisbani et al., 2016) where I appear as co-first author in the publication. I was able to publish a review titled Tau: a common denominator and therapeutic target for neurodegenerative disorders (Maxan et al., 2018) which was published in Journal of Experimental Neuroscience. I was also able to contribute to a manuscript studying the use of a novel monoclonal antibody to target extracellular mHTT (Bartl et al., 2020), which is currently under revisions following a submission to Molecular Therapy. Finally, I am currently contributing to other projects evaluating the presence of tau pathology in grafted HD patients in a collaboration with the University of Washington, as well as another project researching the behavioral phenotypes of a LRP1 x zQ175 murine model of HD.
List of peer-reviewed publications Peer-reviewed publications
Cisbani G*, Maxan A*, Kordower J, Planel E, Freeman T, Cicchetti F. Presence of tau pathology in striatal allografts in Huntington’s and Parkinson’s disease patients. Brain: 140; 2982-2992. 2017. *Equally contributing author
Maxan A, Cicchetti F. Tau: a common denominator and therapeutic target for neurodegenerative disorders. Journal of Experimental Neuroscience, 2018, 12:1179069518772380.
Maxan A, Mason, S, Saint-Pierre M, Smith E, Ho A, Harrower T, Watts C, Tai Y, Pavese N, Savage J, Tremblay M-E, Gould P, Rosser A, Dunnett S, Piccini P, Barker R, Cicchetti F. Outcome of cell suspension allografts in a patient with Huntington’s disease. Annals of Neurology, 2018, 84(6):950-956.
Masnata M*, Sciacca G*, Maxan A*, Denis H, Lauruol F, David L, Saint-Pierre M, Kordower J, Bousset L., Melki R., Alpaugh M, Cicchetti F. Demonstration of prion-like properties of mutant huntingtin fibrils in both in vitro and in vivo paradigms. *Equally contributing authors. Acta Neuropathologica, 2019, 137(6):981-1001. Maxan A, Sciacca G, Alpaugh L, Tao Z, Breger L, Dehay B, Zhang L, Chuan Q, Oueslati A, Bezard E, Cicchetti F. Use of adeno-associated virus-mediated delivery of mutant huntingtin to study the spreading capacity of the protein in mice and non-human primates. Neurobiology of Disease, 2020, 141:104951. doi: 10.1016/j.nbd.2020.104951.
Bartl S, Oueslati A, Southwell A, Siddu A, Parth M, David L, Maxan A, Salhat N, Burkert M, Mairhofer A, Pakevych H, Balazs K, Staffler G, Hayden M, Cicchetti F. The use of monoclonal antibodies to target extracellular mutant huntingtin: implications for the treatment of Huntington’s disease. Neurobiology of Disease, 2020, 141:104943. doi: 10.1016/j.nbd.2020.104943.
Gosset P*, Maxan A*, Alpaugh M, Breger L, Dehay B, Tao Z, Zhang L, Qin C, Cisbani G, Fortin N, Lacroix S, Oueslati A, Vonsattel JP, Bezard E, Cicchetti F. Evidence for the spread of human-derived mutant huntingtin protein in mice and non-human primates. Neurobiology of Disease, 2020, 141:104941. doi: 10.1016/j.nbd.2020.104941.
Introduction
1. Huntington’s disease
Huntington’s disease (HD) is a devastating autosomal dominant neurodegenerative disease typically affecting 4-15 out of every 100 000 people of European decent (Conneally, 1984; Ross and Tabrizi, 2011); numbers that can vary geographically (Mason and Barker, 2009). The disease results from a single unstable mutation in the huntingtin (HTT) gene located on the short arm of chromosome 4 at 4p16.3 (Gusella et al., 1983). While everyone harbours the gene, the mutation leads to the expansion of the cytosine-adenine-guanine (CAG) stretch located in exon 1 (Figure 1). When this expansion results in the CAG repeat domain that codes for polyglutamine (polyQ) sequences to be above a critical threshold (>36 repeats), full penetrance of the disease occurs. Partial penetrance can happen when this expansion is 35 or 36 repeats. Longer CAG repeat domains are associated with earlier ages of disease onset (Foroud et al., 1999; Gusella and MacDonald, 2000; Squitieri et al., 2001) and sequences of up to 200 repeats have been previously reported (Nance et al., 1999). In such cases, the symptoms manifest quite early in life (<20 years of age) in a form that is referred to a Juvenile HD.
Figure 1. The genetics of HD (From: https://ghr.nlm.nih.gov/condition/huntington-disease, the National Library of Medicine). 2. Symptomatology
HD is characterized by an amalgamation of motor dysfunctions, cognitive impairments and psychiatric disturbances. Symptoms typically become apparent between the ages of 35 and 50 (Duyao et al., 1993; Foroud et al., 1999) and progress in severity until death, which occurs in the subsequent decade or two (Conneally, 1984). Motor dysfunctions usually manifest as chorea; short involuntary movements that can be persistent, although other motor irregularities such as dystonia, akinesia, bradykinesia and rigidity become more and more
predominant as the disease progresses (Phillips et al., 2008). Adding to motor impairments, cognitive symptoms can arise at later stages of the disease (Papp et al., 2011; Mestre and Ferreira, 2012; Peavy et al., 2010; Phillips et al., 2008). Additionally, psychiatric symptoms are common and evident in forms of mood disorders, depression, psychosis and can, in some cases, lead to suicide (Shoulson and Young, 2011). Throughout disease progression, constant care becomes a necessity as patients begin to lose their autonomy in everyday activities.
3. Neuropathology
Neuropathologically, HD is characterized by a severe atrophy of the neostriatum resulting from a remarkable neuronal loss, which initially takes place in the caudate nucleus to subsequently involve the putamen (Mann et al., 1993; Myers et al., 1991; Vonsattel and DiFiglia, 1998; Vonsattel et al., 1985). This striatal neuronal degeneration occurs along a caudo-rostral and dorso-lateral/medio-ventral axis (Vonsattel and DiFiglia, 1998). Medium spiny neurons (MSNs) found in the striatum are the primary targets of the neuronal degeneration that occurs during disease progression. Surviving MSNs are often found to present with dendritic changes such as an altered shape and size as well as diminishing dendritic spines (Vonsattel and DiFiglia, 1998; Vonsattel, 2008). While all MSNs are GABAergic neurons, they differ among themselves in their expression of neuropeptides and their projection sites (Gerfen, 1992). During early stages of the disease, GABAergic neurons expressing enkephalin (ENK) and D2 receptors that project to the external segment of the globus pallidus (Gpe) are primarily affected (Gerfen, 1992). In more advanced stages, the MSNs that express substance P and D1 receptors, and that project to the internal segment of the globus pallidus (Gpi) and the substantia nigra pars compacta, are in turn targeted (Reiner et al., 2011; Richfield et al., 1995; Sieradzan and Mann, 2001). Striatal interneurons, which are estimated to represent 5% of the striatal cell population (Sieradzan and Mann, 2001; Steiner and Tseng, 2010) are largely spared (Cicchetti and Parent, 1996; Cicchetti et al., 2000; Sieradzan and Mann, 2001; Wu and Parent, 2000). However, due to the significant atrophy observed, it has been reported that at the final stages of disease, both spiny and aspiny neurons may eventually be vulnerable to pathological processes (Vonsattel, 2008).
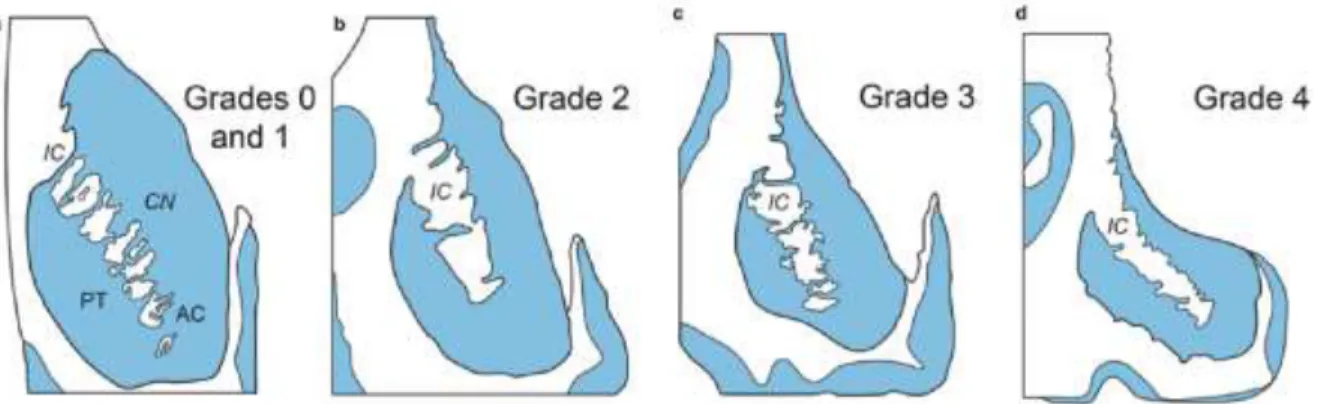
As the disease progresses, various structures of the brain impacted by neuronal degeneration become atrophied. This striking feature has in fact been used to classify the stages of disease progression. The elaborate grading scale (Grades 0 to 4) is based on the macroscopic examination of brain sections, which is used to assess the severity of brain atrophy, while microscopic evaluation helps gauge the extent of neuronal loss and astrogliosis (Vonsattel et al., 1985) (Further described in legend of Figure 2). It should be noted that greater atrophy and neuronal loss have also been associated to longer trinucleotide repeats (Furtado et al., 1996).
Figure 2. Progressive degeneration of the striatum in HD. (a–d) Schematized frontal sections through the rostral level of the striatum
with the head of the caudate nucleus (CN), putamen (PT), and nucleus accumbens (AC) (level CAP). (a) Grades 0 and 1 of striatal atrophy in Huntington’s disease (HD): normal gross aspect of the neostriatum (CN and PT) and AC. (b) Grade 2 of striatal atrophy in HD: markedly atrophic head of the CN. The convex outline of the ventricular surface of the CN, however, is still retained. The PT already shows an obvious volume loss, while the AC appears macroscopically normal. (c) Grade 3 of striatal atrophy in HD: moderate to severe atrophy of the neostriatum. The medial outline of the CN is now flat and forms a nearly straight-line configuration, which parallels the anterior limb of the internal capsule (IC). (d) Grade 4 of striatal atrophy in HD: very severe atrophic CN and PT with markedly concave medial outline of the head of the CN and IC. The AC of grade 4 individuals is also atrophic (Modified according to Vonsattel et al. 1985). No evidence of brain atrophy at gross examination can be found in individuals at Grade 0 – which corresponds to those classified as pre-symptomatic. Measurable atrophy is still not observed by Grade 1, however nearly 50% of neurons of the head of the caudate nucleus have died at this stage. This is further accompanied by a moderate fibrillary gliosis. Atrophy of the caudate nucleus becomes evident at Grade 2. Despite no indications of ventricular enlargement, the putamen may also show signs of atrophy at this stage. Microscopically, neuronal loss, accompanied by fibrillary gliosis, is observed both in the head of the caudate nucleus and in the dorsal putamen. Grade 3 is characterized by a severe atrophy of the caudate nucleus. The putamen is also moderately atrophied, but the shape of the ventricles appears to flatten. Significant neuronal loss and astrogliosis of both the caudate nucleus and the putamen are observed at this stage. In the final stages of the disease, referred to as Grade 4, both the caudate nucleus and putamen are atrophied, leaving the ventricular surface concave. Nearly 95% of neurons have degenerated at this stage (Vonsattel et al., 1985) and severe astrogliosis throughout the neostriatum is present. Additionally, atrophy of other brain structures such as the GP, neocortex and thalamus is notable in Grades 3 and 4, while absent or extremely mild in Grade 1 and 2 patients (Vonsattel, 2008).
Neuronal degeneration can greatly affect the cortex as well. In humans, the cortical mantel is divided into six layers. Layers III, V and VI all contain copius cortical pyramidal neurons (CPNs). The CPNs from layers III and V are the primary input to the basal ganglia, brain stem and spinal cord (Bunner & Rebec, 2016), while the CPNs from layer VI project to the thalamus (McGeorge and Faull, 1989; Shipp, 2007; Estrada-Sánchez and Rebec, 2013). Previous studies conducted on post-mortem tissue obtained from HD patients have reported a 30% reduction in CPNs within these three specific cortical layers (Cudkowicz and Kowall, 1990; Hedreen et al., 1991; Sotrel et al., 1991; Heinsen et al., 1994).
Brain atrophy is not the only neuropathological feature commonly associated with HD. Heightened microglial activation has been reported in the brains of HD patients. While the number of microglial cells is modestly increased between Grades 0 and 3, their density is noticeably higher in the brains of Grade 4 patients (Maxan et al., 2019; Myers et al., 1991; Vonsattel et al., 1985). This microglial response measured by 11CI-PK11195
PET imaging has been suggested to take place early in disease progression (Pavese et al., 2006; Tai et al., 2007) and to further correlate with the severity of the pathology (Sapp et al., 2001).
4. Proteins in neurodegenerative disorders
A finding common to many neurodegenerative diseases is the presence of misfolded protein aggregates, including amyloid-β in Alzheimer’s disease (AD) and α-syn in Parkinson’s disease (PD), in addition to mHTT in HD. This commonality presents a potential target for therapeutic intervention. Previous studies have addressed mHTT aggregation by elevating expression of Heat-Shock Proteins (HSPs) (Fujimoto et al., 2005), promoting clearance of mHTT aggregates (Tanaka et al., 2004) and by polyglutamine peptide inhibitors (Nagai et al., 2003). No compounds targeting aggregation have been approved for human treatment. In this thesis, I will take this shared feature of protein aggregation, and examine if disease toxicity differs when intermediates in the aggregation process are isolated and introduced into the CNS healthy subject. In order to do so, we first need to cover the specifics of the protein, its endogenous functions and the aggregation process.
5. The normal form of the HTT protein
HTT is a cytoplasmic protein that is ubiquitously expressed and present both in humans and rodents, with high expression levels in the brain, cardiovascular system, skeleton, digestive tract and in genital organs (Sassone et al., 2009). It is associated with several organelles, microtubules and vesicular membranes, suggesting a role of the protein in intracellular trafficking, exocytosis and endocytosis (DiFiglia et al., 1995; Gutekunst et al., 1995). HTT is also associated with proteins involved in synaptic functions (Steffan et al., 2000). It presents anti-apoptotic properties (Cattaneo et al., 2005) and plays a critical role in embryonic development (White et al., 1997). Evidence has suggested that a minimum of 30-40% expression of non-mutated HTT is required to support normal development (Gagnon et al., 2010). If overexpressed in certain models (cultured striatal cells, primary cultures from HD mouse models) or in vivo, normal HTT demonstrates protective properties against apoptosis and excitotoxicity (Cattaneo et al., 2005; Leavitt et al., 2006; Zhang et al., 2003). Structurally, the protein is quite large at 350 kDa (Schulte and Troy Littleton, 2011). All orthologs contain a Huntingtin Elongator Factor 3, PR65/A regulatory subunit of PP2A and Tor1 (HEAT) repeats (Tartari et al., 2008; Zhang et al., 2009). This repeat sequence is thought to mediate protein-protein interactions. It has been proposed that the distribution of these repeats within the HTT protein creates a scaffold for forming protein complexes (Takano et al., 2002). Given the large size of the protein, and the presence of a polyQ tract which is thought to reduce solubility (Fiumara et al., 2010), HTT is surprisingly soluble. This is thought to be due to the polyproline (4ngoi) domain that flanks the polyQ tract in higher vertebrates, maintaining the solubility of the protein (Ignatova and Gierasch, 2006) and mediating interactions with vesicle-associated proteins (Qin et al., 2004).
6. The mutated form
In patients with HD, a mutation takes place and an abnormal HTT protein is synthesized. Proteolytic cleavage by calpains, caspases and metalloproteases (Sieradzan et al., 1999; Tarlac and Storey, 2003) release the polyQ stretch which is found at the N-terminal of the protein. It corresponds to approximately 3% of the entire protein (DiFiglia, 2002). Cleaved polyQ stretches are highly prone to form aggregates, as observed in brain sections immunostained for the polyQ domain of the protein (DiFiglia et al., 1995; 1997). They may be found in the cytosol as soluble proteins that can further form oligomers and fibrillary structures (Scherzinger et al., 1997; Zuccato et al., 2010). Unlike in normal individuals, where the HTT protein is found primarily in the cytoplasm, mHTT aggregates are present in both the nucleus and cytoplasm (Kaytor et al., 2004; Poirier et al., 2005) (Johri and Beal, 2010; Steffan et al., 2000). This can cause interference with various cellular pathways associated with the HTT protein such as transcription, axonal transport, energy metabolism, synaptic transmission and vesicle release (Krainc, 2010; Soulet and Cicchetti, 2011), as mentioned above.
Fourier Transformed Infrared Spectroscopy (FTIR) analysis of purified mHTT fragments reveal that shorter polyQ stretches of the protein tend to adopt a β-structure (βstrand/β-turn), whereas longer polyQ chains yield more globular structures, which likely have biological relevance (Monsellier et al., 2015). Globular intermediates can form protofibrils, which ultimately lead to mature fibrils (Figure 3). Conformational changes are important indicators of protein toxicity (Poirier et al., 2002). Observations collected in a model of mHTT aggregation, developed in transiently transfected COS-7 cells, have led to the idea of a dynamic four-step process to explain protein aggregation (Ossato et al., 2010). The first is the accumulation phase during which only soluble misfolded monomers are present at low concentrations. In the second phase, small oligomers are formed and remain in equilibrium with monomers. During the third phase, nucleation is triggered at one or more cytoplasmic sites (e.g. nucleation centres), ultimately leading to the formation of larger insoluble inclusions, the fourth phase of aggregation (Ossato et al., 2010). Longer polyQ stretches correlate with faster aggregation rates (Lunkes and Mandel, 1998). It should be noted that normal HTT can also contribute to the formation of aggregates (Rajan et al., 2001). Indeed, shorter polyQ stretches are sequestered in mHTT inclusions. Thus, the cells are impoverished of the wild type (WT) protein which normally exerts beneficial functions (e.g. anti-apoptotic), leading to loss of function.
Figure 3. A schematic model for HTT aggregation. A native monomer can sample a variety of distinct, misfolded monomer conformations, with the relative number and stability of these conformers potentially being polyglutamine (polyQ) length-dependent. Some misfolded monomers likely lead to aggregates, such as annular aggregates, that likely are off-pathway to fibril formation. There appear to be two generic aggregation pathways toward the formation of fibrils structures. (A) One of these pathways proceeds through oligomeric intermediates, some of which may be facilitated by Nt17. The size and stability of oligomeric aggregates can vary widely. A major structural transition must occur within an oligomer to initiate fibril elongation. (B) The other pathway to fibrils is a direct monomer to fibril transition. Oligomerization can also lead to the formation of large amorphous aggregates. All of these higher order aggregates may accumulate together to form the large inclusions that are a hallmark of Huntington’s disease (HD). (From: Arndt et al., 2015; ncbi.nlm.nih.gov/pubmed/25741791)
Aggregate accumulation is dependent on the turnover of the monomeric mHTT and can thus take place over several years, often preceding the symptomatic manifestation of the pathology. The role of aggregate formation and its potential toxicity are still controversial (Kaytor et al., 2004; Poirier et al., 2005). Another disputed aspect is whether aggregates, or their soluble forms such as small dimers or oligomers, are the actual cause of cell death (Saudou et al., 1998). Indeed, not all evidence supports a detrimental role of aggregates and emerging data suggests a neuroprotective role. For example, it has been shown that neurons die in a dose-dependent manner according to the amount of soluble polyQ present within cells. Indeed, the presence of aggregates have also been reported to increase the cell’s life-span (Arrasate et al., 2004). Finally, it is still unclear whether the toxicity of aggregates originates from their intra-molecular interactions or from their intra-cellular localization, and whether the mutation leads to a gain of toxic function due to the presence of an expanded polyQ stretch or
a loss of the beneficial properties of the normal protein, or a combination of both (Atwal et al., 2007; Dragatsis et al., 2000; Zuchner and Brundin, 2008).
7. Gain vs. loss of function
HD pathology is quite unconventional in that it is modeled by both a gain of function by the mutated form of the protein, and a loss of function by the endogenous WT protein. This gain of function by the mutated form, and loss of function of the endogenous from HD mouse models strongly suggest a predominant gain of function. The mere presence of the CAG sequence is toxic. One study expressing the expanded sequence alone caused neurological impairments in mice (Cattaneo et al., 2001). Another study generated YAC128 -/- mice, that express mHTT without the WT form of the protein. The histopathological and behavioral phenotypes of these generated mice were compared with those of YAC128+/+ mice that express the same level of mHTT with normal levels of WT HTT (Raamsdonk et al., 2005). Both mice showed decreased striatal volume and neuronal counts compared to controls, with no difference between groups (van Raamsdonk et al., 2005). As I previously mentioned, HTT is often cleaved by several caspases. Toxic N-terminal fragments have been reported in the brains of YAC72 mice, fragments that formed several months prior to onset of behavioural or neuropathological changes and even resembled the fragments found in post-mortem HD patient samples (Wellington et al., 2002) suggesting a causal role of the fragment in disease progression. Building on this finding, it has been reported that nuclear localization of the fragments occurred earliest and mostly in the striatum. Further evidence was provided supporting a toxic gain of function by showing that the CAG repeat number was inversely proportional to age of disease onset in human patients (Imarisio et al., 2008), especially evident in juvenile cases with a high CAG repeat.
While a loss of any of the functions of HTT could lead to reduced neuroprotection, it is much less clear whether a loss of function in HD is causative or the result of the disease. A dissociation between the neurons that have been found to contain aggregates of the mutant form of the protein, and those that have been described as most vulnerable in the disease (Arrasate et al., 2004), strongly suggest the likelihood of a contribution from a loss of function by the protein. To clarify this, studies genetically altered WT levels and compared them to those of HD animal models. Conditional inactivation of WT HTT in mice has previously shown a similarity between the loss of HTT function and the phenotypes of HD mouse models (Dragatsis, Levine and Zeitlin, 2000). The mice with WT HTT knocked-out showed progressive motor and cognitive deficits, in addition to decreased longevity. These findings were similar to HD mice. Tissue degeneration in both the striatum and cortex were also seen in these mice, as well as the HD mice (Dragatsis, Levine and Zeitlin, 2000). Lastly, the argument brings us back to the neuronal inclusions often associated with the disease. As mentioned previously, age of disease onset and the amount of cortical inclusions both directly correlate with the CAG repeat expansion length (Becher et al., 1998). That said, the neurons that harbour these inclusions do not appear to be most vulnerable to
neurodegeneration. It has been reported that interneurons with high numbers of aggregates are spared from neurodegeneration, suggesting it may be a mere marker rather than cause of disease (Kuemmerle et al., 1999). This finding suggests a strong dissociation between toxicity and the neuronal inclusions, supporting the contribution to pathogenesis from a loss of function, highlighting once again the importance of examining aggregate-intermediate species for their role in the disease pathology.
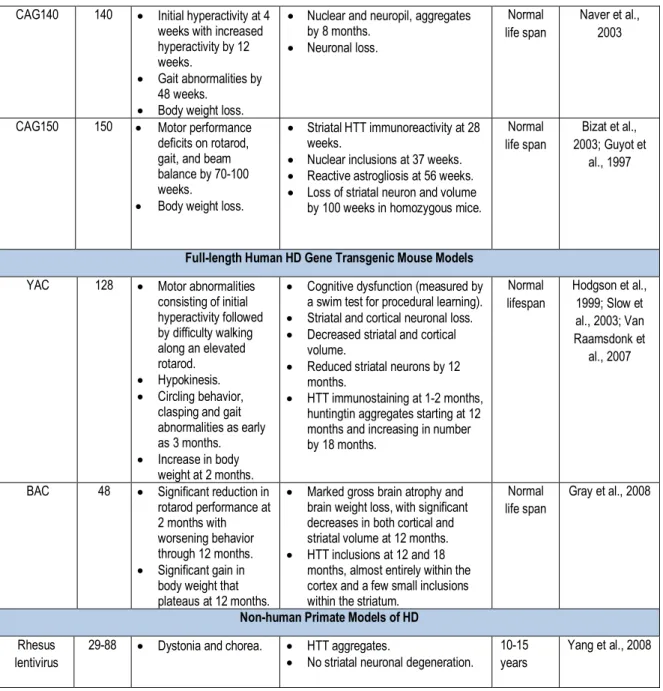
8. Animal Models
To be able to study this protein and how it behaves in living organisms at different timepoints, a number of animal models must be used to cover all aspects of the disease. Most animal models of HD fall into two broad categories: nongenetic and genetic. Historically, nongenetic models have dominated the field of HD research. Although George Huntington first described HD in 1872, researchers did not identify the actual genetic mutation responsible for the disease until 1993, which delayed the development of appropriate genetic models until the last decade. Quinolinic acid (QA) and kainic acid (KA) have been the two most commonly used excitotoxic agents to generate features related to HD in both rodent and non-human primate models. These amino acids (QA and KA) induce cell death by binding to their cognate receptors, N-methyl-D-aspartic acid (NMDA) and non-NMDA, located on striatal neurons. The mitochondrial toxins 3-nitropropionic acid (3-NP) and malonic acid (MA) have also been used in both rodents and non-human primates to produce cell death of striatal neurons via inhibition of the Complex II (succinate dehydrogenase) of the tricarboxylic acid cycle and the electron transport chain in mitochondria, effectively reducing production of adenosine triphosphate (ATP).
More recently created mouse and rat lines express either a truncated or full-length form of the mHTT gene inserted either randomly into the genome (transgenic models) or specifically into the rodent HTT gene locus (knock-in models). The R6/1 and R6/2 transgenic mouse models were first characterized by Bates and colleagues (Mangiarini et al. 1996) and are still the most widely used transgenic mouse models. These mice express mutant exon 1 of the human HTT gene and exhibit both early and severe behavioral and anatomical features of the disease. R6/1 mice express 114 CAG repeats and R6/2 mice, 150 repeats. N171-82Q transgenic mice created by Borchelt and colleagues express the first 171 amino acids of the HTT protein bearing 82 CAG repeats (Schilling et al. 1999). Behavioral and anatomical impairments in this model develop over a more protracted time course than those of the R6 models. The creation of yeast artificial chromosome (YAC) transgenic mice involves cloning an artificial yeast vector that contains an expanded polyglutamine repeat into the mouse genome (Hodgson et al. 1996), and the establishment of a transgenic rat model involves a similar process (von Horsten et al. 2003). These rats express 51 CAG repeats and display both behavioral and anatomical deficits. Rats, in general, tend to live approximately 1 year longer than mice and have a more complex behavioral repertoire, making the transgenic rat model an attractive candidate for thorough, long-term
therapeutic studies. Knock-in models that use gene targeting to insert expanded CAG repeat coding regions into the mouse HTT locus have also recently been created. One new promising transgenic mouse has been created by using a bacterial artificial chromosome (BAC) that contains the entire 170 kb human HTT locus. These mice are characterized by progressive motor deficits and can show significant reduction in performance on the rotarod as early as 2 months of age which worsen until 12 months. Notably, while the BACHD model expresses the entire gene and even contains 97 glutamine repeats, the BACHD model does not typically express mHTT aggregates (Pouladi et al., 2012). The properties of the remaining animal models of HD are further summarized in Table 1 while animal models using recombinant vectors are summarized in Chapter III.
N-terminal Fragment Models of HD Model CAG
repeats
Behavioral Changes Neuropathology Survival References
R6/1 mouse 116 • Motor performance abnormalities at 4-5 months.
• Significant body weight loss at 22 weeks.
• Reduced brain volume by 18 weeks.
• Neuronal atrophy but not neuronal loss.
• HTT aggregates at 2 months. Reduced dopamine levels.
12 months Sun et al., 2002; Hodgson et al., 1996 R6/2 mouse 144-150
• Dystonia with limb clasping by 6 weeks. • Reduced hindlimb and
forelimb coordination by 9 and 11 weeks, respectively. • Rotarod changes by 5 weeks. • Seizures.
• Body weight loss by 8-9 weeks.
• Significant brain weight loss at 30 days.
• Gross brain atrophy by 60 days with hyperventricular enlargement. • Progressive neuronal atrophy with
neuronal loss by 90 days. • Progressive HTT aggregate
formation starting at day 1. • Astrogliosis at 90 days. • Reduced dopamine levels.
12-18 weeks Sun et al., 2002; Laforet et al., 2001 N171-82Q mouse 82 • Motor performance deficits, and limb clasping by 11 weeks. • Water maze deficits at
14 weeks. • Body weight loss.
• Gross brain atrophy. • Hyperventricular enlargement. • Striatal neuron atrophy and loss. • HTT aggregates by 16 weeks. 130-180 days Blum et al., 2002; Rebec et al., 2002; Bolivar et al., 2003; Stack et al., 2007 Transgenic rat 51 • Progressive rotarod decline by 2 months. • Significant body weight
loss by 24 months. • Cognitive deficits by 12 months. • Head dyskinesias by 10 months. • Hyperventricular enlargement. • HTT inclusions.
• Striatal neuron loss.
None reported
Petersen et al., 2002; Bogdanov
et al., 1998
Knock-in Mouse Models of HD
Hdh/Q72-80
72-80 • Aggressive behavior. • Rotarod impairment. • No weight loss.
• No neuronal loss or reactive astrogliosis.
• HTT aggregates by 28 weeks with nuclear aggregates by 96 weeks.
Normal life span Alexi et al., 1998 HdhQ111 109-111 • Gait impairments by
96 weeks. • Nuclear inclusions by 48 weeks • Neuropil aggregates by 68 weeks.
Normal life span
Brouillet et al., 1998
CAG140 140 • Initial hyperactivity at 4 weeks with increased hyperactivity by 12 weeks.
• Gait abnormalities by 48 weeks.
• Body weight loss.
• Nuclear and neuropil, aggregates by 8 months. • Neuronal loss. Normal life span Naver et al., 2003
CAG150 150 • Motor performance deficits on rotarod, gait, and beam balance by 70-100 weeks.
• Body weight loss.
• Striatal HTT immunoreactivity at 28 weeks.
• Nuclear inclusions at 37 weeks. • Reactive astrogliosis at 56 weeks. • Loss of striatal neuron and volume
by 100 weeks in homozygous mice.
Normal life span
Bizat et al., 2003; Guyot et
al., 1997
Full-length Human HD Gene Transgenic Mouse Models
YAC 128 • Motor abnormalities consisting of initial hyperactivity followed by difficulty walking along an elevated rotarod. • Hypokinesis. • Circling behavior,
clasping and gait abnormalities as early as 3 months. • Increase in body
weight at 2 months.
• Cognitive dysfunction (measured by a swim test for procedural learning). • Striatal and cortical neuronal loss. • Decreased striatal and cortical
volume.
• Reduced striatal neurons by 12 months.
• HTT immunostaining at 1-2 months, huntingtin aggregates starting at 12 months and increasing in number by 18 months. Normal lifespan Hodgson et al., 1999; Slow et al., 2003; Van Raamsdonk et al., 2007
BAC 48 • Significant reduction in rotarod performance at 2 months with worsening behavior through 12 months. • Significant gain in
body weight that plateaus at 12 months.
• Marked gross brain atrophy and brain weight loss, with significant decreases in both cortical and striatal volume at 12 months. • HTT inclusions at 12 and 18
months, almost entirely within the cortex and a few small inclusions within the striatum.
Normal life span
Gray et al., 2008
Non-human Primate Models of HD
Rhesus lentivirus
29-88 • Dystonia and chorea. • HTT aggregates.
• No striatal neuronal degeneration. 10-15 years
Yang et al., 2008
Table 1. Animal models of HD. Adapted from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2693467/. 9. Transcellular propagation
It has recently been described that mHTT aggregates have been reported within intracerebral allografts of striatal tissue in 3 HD patients who received foetal allograft transplants approximately 10 years earlier and then died secondary to the progression of their disease. The mHTT+ aggregates were observed in the extracellular matrix of the transplanted tissue, whereas in the host brain they were seen in neurons, neuropil, extracellular matrix and blood vessels (Cicchetti et al., 2014). This was the first demonstration of the presence of mHTT in genetically normal and unrelated allografted tissue transplanted into the brain of an HD patient. These results raised questions on protein spread in HD, and whether mHTT may propagate between cells. Several in vitro
studies have also suggested that mHTT may potentially propagate from cell to cell (Costanzo et al., 2013; Ren et al., 2009; Herrera et al., 2011; Yang et al., 2002), suggesting this process may be characteristic of the disease and could be demonstrated in vivo as well.
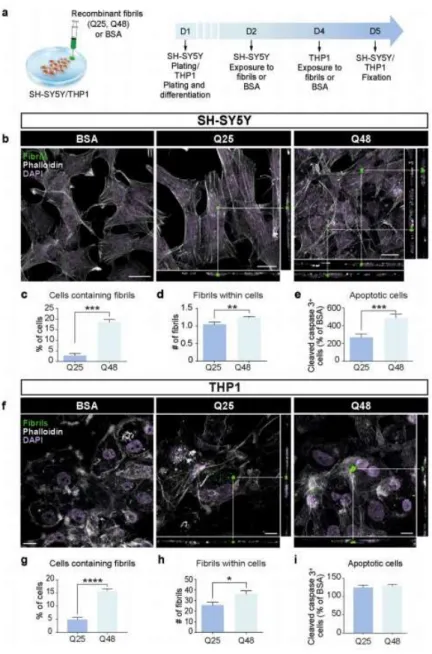
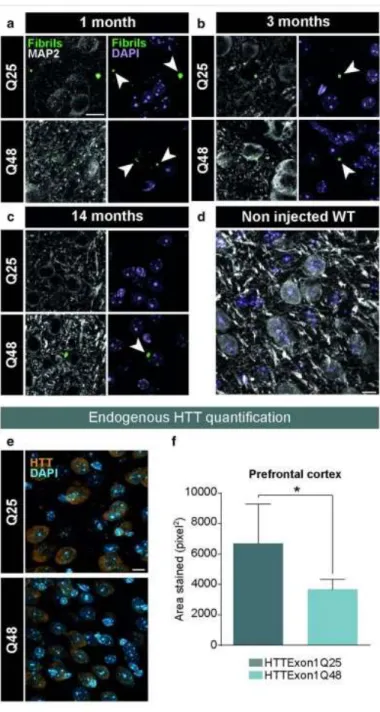
Throughout the last decade it was determined that various cell types, including neuronal-like cells, were capable of internalizing synthetic mHTT aggregates exogenously delivered in the culture milieu (Ren et al., 2009; Yang et al., 2002). In these experiments, aggregates were localized within the cytoplasm, all were associated with proteins involved in the quality control of the cell such as ubiquitin (Ren et al., 2009). When the synthetic protein was tagged with a sequence mediating its import into the nuclear compartment (nuclear localization signal), aggregates were translocated into the nucleus, preceding cell death (Yang et al., 2002). These observations led to the conclusion that membranes are permeable to large fibrillar structures (Ren et al., 2009; Yang et al., 2002). A Biomolecular Fluorescence Complementation (BiFC)-based assay was developed using non-fluorescent halves of the Venus protein tagged to mHTT. A fluorescent signal was detected when mHTT had become oligomerized. To evaluate whether mHTT was able to propagate between cells, the two halves were expressed independently in separate cell populations and then mixed together. The mixed population presented a significant number of fluorescent cells, which demonstrated the capacity of the protein to migrate from cell to cell and act as a seed for aggregation (Herrera et al., 2011). Other evidence demonstrated that oligomers are toxic and that inclusion bodies are not necessary and neither sufficient to trigger cell death (Herrera et al., 2011; Sassone et al., 2009). mHTT spreading has been found in three different neural network models: human neurons of organotypic brain slices of HD mouse models, an ex vivo corticostriatal slice model and the corticostriatal pathway in vivo (Pecho-Vrieseling et al., 2014). Moreover, healthy human neurons in HD mouse model brain slices displayed non-cell autonomous changes in morphological integrity that were more pronounced when these neurons bore mHTT aggregates (Pecho-Vrieseling et al., 2014). Recent reports on the propagation of mHTT was shown in small rodents when fibroblasts that had been derived from HD patients as well as induced pluripotent stem cells (iPSCs) characterized by 143 CAG repeats were capable of transferring mHTT protein aggregates into healthy, genetically unrelated tissue through implantation in the cerebral ventricles of neonatal mice (Jeon et al., 2016). The effects of exogenous human fibrillar mHTT (Q48) and HTT (Q25) N-terminal fragments have also been tested in three cellular models and three distinct animal paradigms. Two models of human neuronal cells, human macrophages, Wild-type (WT) animals and transgenic pups were incubated or injected with recombinant fibrils. These were found to be taken up by all cultured cell types, inducing morphological changes and death in vitro. In vivo, the injections resulted in cognitive deficits and increased anxiety-like behavior, with post mortem analysis indicating changes in staining patterns of endogenous HTT (Masnata et al., 2019).
The idea that proteins associated to certain diseases can act as prions emerged from autopsy reports (Li et al., 2008; Kordower et al., 2008; Li et al., 2010). Post-mortem analyses of the brains of individuals who suffered from Parkinson’s disease revealed that the young, healthy dopaminergic neurons implanted in the striatum of the patients presented with Lewy body (LB) pathology. It was proposed that prion-like transfer of α-syn could support disease pathology (Li et al, 2008; Brundin et al., 2008). The presence of extracellular α-syn in human plasma and cerebrospinal fluid (CSF) had previously suggested that the protein could enter cells from extracellular space (El-Agnaf et al, 2003). Subsequently, in vitro studies demonstrated that α-syn can be transferred from cell-to-cell (Desplats et al., 2009; Hansen et al., 2011; Kordower et al., 2011; Luk et al., 2012b; Mougenot et al., 2012; Luk et al. 2012a; Ulusoy et al., 2013; Recasens et al., 2014; Holmqvist et al., 2014; Peelaerts et al., 2015; Paumier et al., 2015; Helwig et al., 2016; Koller et al., 2017; Ulusoy et al., 2017; Abdelmotilib et al., 2017). Similarly, it has been proposed that mHTT can act as a prion once phagocytosed by microglia cells in a HD Drosophila model (Pearce et al., 2015). The criteria for which α-syn can be considered as a prion-like protein has been extensively discussed (Dunning et al., 2013) and this criteria can be applied to mHTT as well: 1) it acquires different conformations, forming fibrils and oligomers rich in β-sheets; 2) it can be transferred between cells, although the mechanisms are still largely unexplored; 3) aggregates can propagate
in vivo, both in a murine model as well as in a Drosophila model of HD; and 4) it can recruit the normal protein
into the aggregate leading to a conformational change.
There is now a growing body of evidence that non-cell autonomous mechanisms are involved in protein spread in several neurodegenerative disorders (Polymenidou and Cleveland, 2011). For example, α-syn can be released and taken up by neurons (Desplats et al., 2009; Hansen et al., 2011), and seed pathology both
in vitro and in vivo. Indeed, intracerebral inoculation of brain homogenates derived from old α-syn transgenic
(Tg) mice, or injection of synthetic α-syn preformed fibrils accelerates the formation of α-syn protein aggregates and precipitates neurological dysfunction in animals (Luk et al., 2012; Luk et al., 2012). Further experiments now support the idea that pathological misfolded proteins, such as amyloid (A), tau and -syn retain the capability of propagating in a prion-like fashion (Soto et al., 2012). For example, A diffuse extra-cellularly and was found within WT neuronal grafts in a transplanted AD mouse model (Meyer-Luehmann et al., 2003). Inoculation of brain extracts from AD patients or from aged AD mice into a transgenic model expressing the human amyloid precursor protein increased and accelerated the brain burden in A (Kane et al., 2000; Meyer-Luehmann et al., 2006). Similarly, implantation of murine brain extracts expressing the human tau isoform into the hippocampus of transgenic mice expressingthe longer tau isoform without presenting inclusions promoted the spreading of aggregation from the site of implantation to neighbouring regions (Clavaguera et al., 2009). The observation of LB in transplanted tissue in PD patients (Kordower et al., 2008; Li et al., 2008) further supported the idea that the protein -syn has the capacity to propagate throughout the brain. The observations made in the human PD
transplanted brain were subsequently replicated in a mouse model. Transgenic mice expressing human -syn were transplanted with Green Fluorescent Protein-positive (GFP+) cortical neuronal stem cells in the hippocampus. The number of grafted cells containing inclusion bodies increased over time (Desplats et al., 2009). Furthermore, inoculations of brain extracts from aged -syn expressing mice seeded aggregation in the recipient mouse and propagate in the brain regions via anatomically connected structures (Luk et al., 2012). The propagation properties of -syn, A and tau have all been confirmed in vitro. The proteins can be taken up by neuronal cells and have detrimental consequences. Cells exposed to neuron-derived -syn presented with signs of apoptosis, such as nuclear fragmentation as well as caspase 3 activation, both in vitro and in vivo (Desplats et al., 2009; Danzer et al., 2011). Overall, these observations indicate a prion-like spread of pathological proteins that could contribute to neurodegenerative processes. Pathological proteins retain the ability to propagate from cell to cell, likely sharing some properties of the infectious misfolded prion proteins (Goedert et al., 2010). Importantly, this process may be common to all sporadic neurodegenerative disorders (Soto et al., 2012; Olanow and Prusiner, 2009; De Calignon et al., 2012; Meyer-Luehmann et al., 2003; Bachoud-Lévi et al., 2000; Rosser et al., 2002), although the mechanisms by which transcellular propagation occurs is unclear.
10. Trans-synaptic propagation
Transynaptic transmission of mHTT from cortical projection neurons to grafted tissue may be one major and also most studied routes of disease spread. This hypothesis is in part supported by the abundant expression of mHTT in the layer V of the cortex (Fusco et al., 1999) as well as by the evidence of synaptic connections between cortical neurons and grafted cells (Cicchetti et al., 2009). The ability of misfolded proteins to transfer transynaptically has been previously reported in an AD model (De Calignon et al., 2012; Liu et al., 2012). Selective expression of tau in the entorhinal cortex showed the propagation of the protein in other brain regions anatomically linked (De Calignon et al., 2012; Liu et al., 2012), where targeted cells were also reported to degenerate (De Calignon et al., 2012). Similarly, inoculation of brain lysates from aged -syn-expressing mice or -syn preformed fibrils in the brain of recipient mice, accelerated the formation of aggregates and protein spread in different regions. The protein was found in structures remote to the injection sites but which followed the anatomical circuits (Luk et al., 2012).
It is thus plausible that mHTT aggregates are released into the graft through anatomical connections, and more specifically from cortical projections. HTT is indeed associated with axonal transport (Trushina et al., 2004) and synaptic transmission (Sun et al., 2001). Recently, two reports supported the propagation of mHTT in murine and Drosophila models of HD (Pecho-Vrieseling et al., 2014; Babcock and Ganetzky et al., 2015). Transneuronal propagation of mHTT in the corticostriatal pathway in ex vivo and in vivo models of the disease was found to affect neuronal integrity (Pecho-Vrieseling et al., 2014). mHTT was found to spread in three separate neural
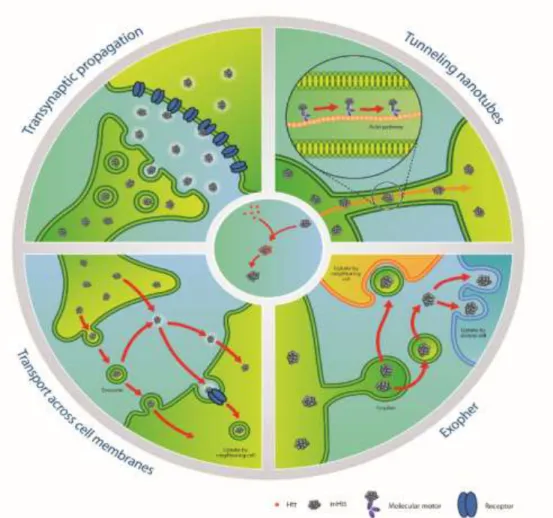
network models: 1) human neurons integrated in the neural network of organotypic brain slices of HD mouse models, 2) an ex vivo corticostriatal slice model and 3) the corticostriatal pathway in vivo. It was also reported that transneuronal mHTT propagation could be blocked by two different botulinum neurotoxins, each known for inactivating critical components of the synaptic vesicle fusion machinery (Pecho-Vrieseling et al., 2014). It was then shown that mHTT accumulates in the synaptic terminals of an HD Drosophila model. This study also demonstrates the capability of mHTT to migrate and diffuse through the brain before being internalized by other neurons, eventually leading to non-cell autonomous degeneration (Babcock and Ganetzky, 2015). Importantly, inhibiting endocytosis in the receiver neurons can prevent degeneration. The expression of mHTT in different neuronal populations appears to lead to different patterns of propagation. These different patterns are summarized in Figure 4.
Figure 4. Putative mechanisms of mHTT spreading and seeding capacities. Left upper panel: illustration of propagation of
mHTT. Right upper panel: transport mechanism of mHTT via tunneling nanotubes. Left lower panel: mHTT can be released within exosomes or in a free form. After extrusion, exosomes carrying mHTT can fuse with the plasma membrane of a neighboring cell. Alternatively, mHTT can escape from the exosomal-vesicle into the extracellular compartment, with the same fate as the counterpart released directly as a free form. Finally, mHTT can be internalized by a recipient cell via receptor-mediated endocytosis or directly penetrate the plasma membrane. Right lower panel: In neurons of C. elegans, mHTT has been shown to be contained within exophers, an entity which resembles mammalian exosomes. Released exophers may be incorporated by adjacent or distant cells or secrete their contents into the milieu. Central panel: schematic of the seeding process of mHTT. The misfolded protein recruits HTT in an elongation