Echange et Corrélation dans la Structure Electronique des Solides, du Silicium à l'Oxyde Cuivreux: Approximation GW et au-delà
234
0
0
Texte intégral
Figure
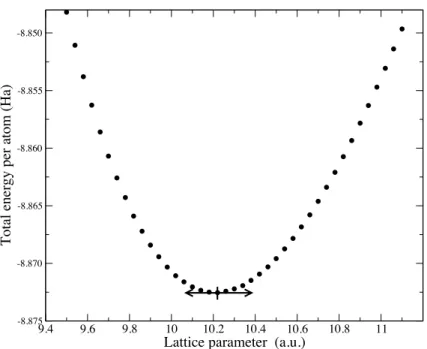
![Figure 4.1: Optical absorption spectrum of bulk silicon calculated within RPA (dotted line), adiabatic LDA (dashed line), and using the α kernel of reference [4] (full line), compared to the experimental curve (full circles) of reference [30]](https://thumb-eu.123doks.com/thumbv2/123doknet/2850095.70498/73.892.261.694.127.451/absorption-spectrum-calculated-adiabatic-reference-compared-experimental-reference.webp)

+7



Documents relatifs