SOLANGE SCHNEIDER
MOLYBDOPHOSPHATE D’AMMONIUM
IMMOBILISÉ SUR SILICE MÉSOPOREUSE POUR
L’ADSORPTION SÉLECTIVE DU RADIOCÉSIUM
Mémoire présenté
à la Faculté des études supérieures et postdoctorales de l’Université Laval dans le cadre du programme de maîtrise en chimie
pour l’obtention du grade de Maître ès sciences (M.Sc.)
DÉPARTEMENT DE CHIMIE FACULTÉ DES SCIENCES ET DE GÉNIE
UNIVERSITÉ LAVAL QUÉBEC
2012
Résumé
Les sources de radiocésium-137 (137Cs) sont utilisées commercialement dans les domaines médicaux et industriels. Il existe plusieurs méthodes, telle la spectroscopie-γ, permettant de détecter de faibles concentrations de 137Cs. Cependant, l’analyse du ratio
135Cs/137Cs, qui permet de caractériser la source, ne peut être obtenue que par spectrométrie
de masse, et seulement si l’élément a été préalablement séparé chimiquement, et ce, pour éviter les interférences isobariques.
Ce mémoire porte sur la séparation du Cs+ par l’utilisation d’adsorbants à base de molybdophosphate d’ammonium (AMP), sel connu pour être sélectif au Cs+, supportés sur silice mésoporeuse (SBA-15). Les silices mésoporeuses permettent d’augmenter la surface de contact avec l’échantillon et de stabiliser l’échangeur de cations pour des perspectives de régénération.
Des analyses ont été faites quant aux caractéristiques chimiques des matériaux par plasma à couplage inductif-spectrométrie de masse, diffraction des rayons X, thermogravimétrie et spectroscopie de photoélectrons induits par rayons X. Les caractéristiques physiques des matériaux ont quant à elles été déterminées par microscopie électronique et par physisorption d’azote à -196 °C. Finalement, les capacités des matériaux ont été évaluées par analyse élémentaire par plasma à couplage inductif-spectrométrie de masse.
Abstract
Radiocesium (137Cs) sources are commercially used in medical and industrial domains. Several methods, including γ-spectroscopy, can detect low concentrations of
137Cs. However, only mass spectroscopy can achieve the determination of the 135Cs/137Cs,
which is important to characterize the radiosource. Radiocesium having several isobaric interferences, it is hence necessary to chemically separate the cesium from its matrix.
Cesium separation has been studied by using ammonium molybdophosphate (AMP) based adsorbents, this salt being known to be selective to Cs+. These sorbents are supported on mesoporous silicas (SBA-15), which allows to enhance the specific surface of the material and thus stabilizes the cationic exchanger, allowing regeneration.
Materials have been analysed by inorganic mass spectrometry, X-ray diffration, thermogravimetric analysis and by X-ray photoelectron spectroscopy to determine their chemicals characteristics. Physical characteristics have been determined by electronic microscopy and by nitrogen physisorption at -196 °C. Extraction capacities have also been studied by inorganic mass spectrometry.
Avant-propos
Je tiens à remercier tout d’abord mes professeurs, messieurs Freddy Kleitz et Dominic Larivière, pour l’aide qu’ils m’ont offerte durant tout le temps nécessaire pour l’exécution de ce projet, de même que M. François Bilodeau, de la centrale Gentilly-II d'Hydro-Québec, qui m’a fait voir l’applicabilité et l’utilité de ce projet. Pour leur aide aussi : messieurs Serge Groleau et Jean Frenette, sans qui certains problèmes n’auraient pas toujours été résolus.
Je voudrais aussi remercier Alexandre, Pablo, Charles, Sabrina, Laurence, Ana, Luc et Annie, ainsi que Rémy, Jean-Luc, Nima et Mahesh, pour m’avoir permis de décrocher de temps en temps, et de discuter et rire de tout et de rien. Sans eux, le temps aurait parfois semblé beaucoup trop long. Merci aussi aux stagiaires qui ont travaillé avec moi, Mélodie, Adriana et Jean-Daniel, qui m’ont donné d’énormes coups de main et qui ont vu, eux aussi, le côté fascinant de ce projet.
Pour finir, j’aimerais remercier Samuel, qui m’a écouté raconter mes problèmes, sans nécessairement les comprendre, mais en m’apportant toujours son point de vue, surtout pour me dire que, finalement, ce n’était que de la science…
L’environnement, c’est tout ce qui n’est pas moi.
Table des matières
Résumé ... I Abstract ... III Liste des figures ... XI Liste des tableaux ... XIII Liste des abréviations et symboles ... XV
1. Problématique ... 1
1.1 Techniques d’analyse du césium ... 10
1.2 Techniques de séparation du césium ... 12
1.2.1 Chromatographie ionique ... 12
1.2.2 Ferrocyanates de métaux alcalins ... 14
1.2.3 Molybdophosphate d’ammonium ... 14
1.2.3.1 Molybdophosphate d’ammonium supporté ... 16
1.2.3.1.1 Supports organiques ... 16
1.2.3.1.2 Supports inorganiques ... 17
1.3 Matériaux poreux et ions de Keggin ... 18
1.4 Silices mésoporeuses ... 19
1.5 Objectifs du projet ... 21
2. Techniques de caractérisation ... 23
2.1 Physisorption de gaz ... 23
2.2 Microscopie électronique et analyse dispersive en énergie ... 26
2.3 Diffraction des rayons X à bas et grands angles ... 27
2.4 Résonance magnétique nucléaire du phosphore-31 à l’état solide ... 27
2.5 Spectroscopie de photoélectrons induits par rayons X ... 29
2.6 Spectrométrie infrarouge à transformée de Fourier ... 29
2.7 Thermogravimétrie et analyse différentielle thermique ... 30
2.8 Spectrométrie de masse couplée à un plasma inductif... 30
3. Section expérimentale ... 33
3.1 Réactifs utilisés... 33
3.2 Synthèse de la SBA-15 ... 34
3.3 Synthèse des matériaux de molybdophosphate d’ammonium sur silice SBA-15 ... 36
3.4 Digestion acide des matériaux ... 37
3.6 Cinétique d’adsorption du césium ... 38
3.7 Adsorption du césium sur les matériaux ... 38
3.8 Profil d’élution du césium ... 39
3.9 Réutilisation du matériau ... 39
3.10 Adsorption des milieux naturels simulés ... 39
3.11 Instrumentation ... 39
4. Synthèse et caractérisation des matériaux ... 42
4.1 Optimisation de la méthode de synthèse... 42
4.2 Physisorption d’azote ... 53
4.3 Microscopie électronique et analyse dispersive en énergie ... 55
4.4 Diffraction des rayons X à bas et grands angles ... 57
4.5 Résonance magnétique nucléaire à l’état solide du phosphore-31 ... 59
4.6 Spectroscopie de photoélectrons induits par rayons X ... 60
4.7 Infrarouge à transformée de Fourier ... 61
4.8 Thermogravimétrie et analyse thermo-différentielle ... 63
4.9 Spectrométrie de masse couplée à un plasma inductif ... 64
5. Efficacité des matériaux ... 66
5.1 Capacités des matériaux hautement chargés ... 66
5.2 Capacité d’adsorption du césium sur les matériaux ... 69
5.3 Élution du césium ... 74
5.4 Adsorption du césium en milieux naturels simulés ... 76
5.5 Matériaux irradiés ... 77
6. Conclusion ... 82
7. Perspectives ... 83
Bibliographie ... 87
Annexe 1 : Diffractogramme des rayons X à grands angles d’un matériau AMP/SBA-15 6 % massique lavé avec NH4Cl ... 95
Annexe 2 : Protocole de synthèse des monolithes ... 96
Liste des figures
Figure 1 : Pourcentages relatifs des produits de désintégration du combustible nucléaire
(Y, %) selon sa nature (A). ... 2
Figure 2 : Quantités en Bq/m2 de césium-137 associé aux essais nucléaires mesurées dans l’hémisphère Nord ... 3
Figure 3 : Nombre d’essais nucléaires atmosphériques et souterrains selon le pays et l’année ... 4
Figure 4 : Principaux modes de dispersion du radiocésium et leur contribution à la quantité totale dans l’environnement ... 6
Figure 5 : Rapport Cs-135/Cs-137 selon la zone d’échantillonnage ... 6
Figure 6 : Principe de datation des sédiments par la mesure du radiocésium selon la profondeur dans la carotte ... 8
Figure 7 : Chaîne de désintégration du césium-137 ... 12
Figure 8 : Exemples d'éther-couronne (gauche) et de calixarène (droite) ... 13
Figure 9 : Structure de Keggin ... 15
Figure 10 : Exemples de structures des silices mésoporeuses : a) p6mm, b) Ia d, c) Im m, d) Fm m ... 20
Figure 11 : Procédé de synthèse des silices mésoporeuses bidimensionnelles hexagonales (ex. SBA-15) ... 20
Figure 12 : Classification des isothermes de physisorption selon l’IUPAC ... 23
Figure 13 : Classification des hystérèses selon l'IUPAC ... 24
Figure 14 : Isotherme d’une SBA-15 expliquée par le processus d'adsorption des molécules d'azote dans un pore ... 25
Figure 15 : Diffraction des rayons X dans un cristal ... 27
Figure 16 : Mouvement de précession d'un noyau en RMN ... 28
Figure 17 : Représentation schématique d'un ICP-MS ... 31
Figure 18 : Programme de calcination de la SBA-15 ... 35
Figure 19 : Processus final de synthèse des matériaux AMP/SBA-15 à partir de H3PMo12O40 ... 37
Figure 20 : Processus de synthèse des matériaux AMP/SBA-15 à partir de Cs2CO3pour des charges de 30 % et 50 % massique en AMP ... 42
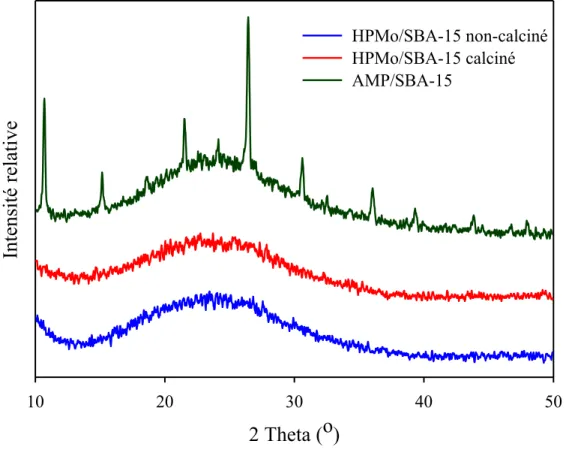
Figure 21 : Diffraction des rayons X pour un matériau AMP/SBA-15 fait à partir de Cs2CO3 comparé à de l’AMP disponible commercialement ... 44
Figure 22 : Processus de synthèse des matériaux AMP/SBA-15 à partir de H3PMo12O40 .. 45
Figure 23 : Diffractogramme des rayons X d’un matériau AMP/SBA -15 chargé à 75 % massique et lavé avec NH4NO3 1 M ... 46
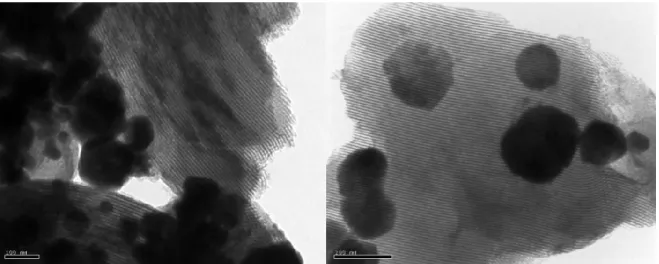
Figure 24 : Images prises en microscopie électronique à transmission d’un matériau chargé à 75 % massique et lavé avec NH4NO3 1 M ... 47
Figure 25 : Diffractogramme des rayons X d’un matériau AMP/SBA -15 chargé à 25 % massique et lavé avec NH4NO3 1 M selon l’étape de synthèse ... 48
Figure 26 : Diffractogramme des rayons X d’un matériau AMP/SBA -15 chargé à 75 % massique et lavé avec NH4NO3 0,4 M ... 49
Figure 27 : Images prises en microscopie électronique à transmission d’un matériau chargé
à 75 % massique et lavé avec NH4NO3 0,4 M ... 50
Figure 28 : Images prises en microscopie électronique à transmission d’un matériau chargé
à 6 % massique et lavé avec NH4NO3 0,4 M ... 51
Figure 29 : Diffractogrammes des rayons X de matériaux calcinés à 350 oC (gauche) et 450 oC (droite) selon la durée ... 52
Figure 30 : Isothermes de physisorption d’azote à -196 °C pour un matériau AMP/SBA-15
chargé à 6 % m/m et la silice SBA-15 utilisée au départ. ... 54
Figure 31 : Images prises en microscopie à transmission pour un matériau AMP/SBA-15
chargé à 6 % massique ... 55
Figure 32 : Analyse dispersive en énergie faite en HR-TEM pour un matériau ... 56 Figure 33 : Diffractogramme des rayons-X à bas angles pour un matériau AMP/SBA-15
chargé à 6 % massique et la SBA-15 de départ ... 57
Figure 34 : Diffractogrammes des rayons X à grands angles de l’AMP commercial et d’un
matériau AMP/SBA-15 chargé à 6 % m/m ... 58
Figure 35 : Spectre RMN à l’état solide du 31P d’un matériau AMP/SBA-15 chargé à 6 % m/m ... 59
Figure 36 : Énergie de l'orbitale O 1s dans un échantillon AMP/SBA-15 6 % m/m ... 60 Figure 37 : Spectres FTIR en absorbance de l’AMP commercial, d’un matériau
AMP/SBA-15 6 % m/m et de SBA-15 de départ ... 62
Figure 38 : Structure cristalline α de l’ion de Keggin et les différents types d’oxygènes
présents dans celui-ci ... 63
Figure 39 : Analyses thermogravimétrique et thermo-différentielle d’un matériau acide
phosphomolybdique/SBA-15 chargé à 6 % ... 64
Figure 40 : Lixiviation sur 6 h d’un matériau AMP/SBA-15 6 % m/m dans HNO3 10 %
v/v, HCl 10 % v/v, H2O et NH4OH pH 9 ... 65
Figure 41 : Capacités d’adsorption du césium d’un matériau AMP/SBA-15 50 % m/m pour
différents rapports molaires Cs/AMP ... 66
Figure 42 : Volume en ml nécessaire pour éluer tout le césium après une adsorption faite à
Cs/AMP=3 pour un matériau AMP/SBA-15 50 % m/m ... 67
Figure 43 : Réutilisation sur 5 cycles d'un matériau AMP/SBA-15 50 % m/m ... 69 Figure 44 : Cinétique d'adsorption du césium sur un matériau AMP/SBA-15 6 % m/m à
divers rapports Cs/AMP ... 70
Figure 45 : Quantités maximales de césium adsorbé en mg selon le rapport Cs/AMP ... 71 Figure 46 : Profil d'élution du césium pour des concentrations de NH4NO3 de 1, 6 et 12 M
à des rapports Cs/AMP = 3 (gauche) et Cs/AMP = 10 (droite) ... 75
Figure 47 : Diffractogrammes des rayons X d’un matériau AMP/SBA-15 50 % m/m avant
et après irradiation avec une source de Cs-137 ... 78
Figure 48 : Lixiviation comparative d'un matériau AMP/SBA-15 50 % m/m avant (haut) et
après (bas) irradiation avec une source de Cs-137. ... 79
Figure 49 : Tests d'adsorption et d'élution pour un matériau AMP/SBA-15 50 % m/m avant
et après irradiation avec une source de Cs-137 ... 80
Figure 50 : Réutilisation d'un matériau AMP/SBA-15 avant (gauche) et après (droite)
irradiation avec une source de Cs-137 ... 81
Figure 51 : Photographie (gauche) et image prise en microscopie à balayage (droite) d’un
Liste des tableaux
Tableau 1 : Temps de demi-vie et mode de désintégration des isotopes de césium
répertoriés ... 1
Tableau 2 : Réactifs entrant dans la composition de l’eau de mer synthétique ... 34
Tableau 3 : Rapports molaires relatifs des réactifs pour la synthèse de la SBA-15 ... 35
Tableau 4 : Paramètres d’utilisation de l’unité à micro-ondes CEM Mars 5 ... 40
Tableau 5 : Paramètres de l’instrument optimisés pour les analyses de 95Mo, 103Rh, 133Cs et 138Ba en ICP-MS ... 41
Tableau 6 : Limites de détection et de quantification en ICP-MS pour le molybdène-95, le césium-133 et le baryum-138 en milieu HNO3 2 % v/v. ... 41
Tableau 7 : Résultats d’imprégnation de la méthode au départ de Cs2CO3 pour des charges de 30 % et 50 % massique en AMP ... 43
Tableau 8 : Résultats d’imprégnation de la méthode au départ de l’acide phosphomolybdique ... 45
Tableau 9 : Rendement de fixation de l’acide phosphomolybdique selon le temps de calcination et la température ... 52
Tableau 10 : Surface spécifique, volume poreux et diamètre des pores pour un matériau AMP/SBA-15 6 % m/m et la silice SBA-15 utilisée au départ ... 55
Tableau 11 : Pics présents en diffraction des rayons X à grands angles pour un matériau AMP/SBA-15 6 % m/m ... 59
Tableau 12 : Quantités relatives d’AMP à la surface de la silice et à l’intérieur de la structure d’AMP selon les intégrations faites en RMN à l’état solide du 31P ... 60
Tableau 13 : Coefficients de distribution et critères correspondants ... 68
Tableau 14 : Quantités maximales de césium adsorbé selon le rapport Cs/AMP pour un matériau AMP/SBA-15 6 % m/m ... 71
Tableau 15 : Résultats d’adsorption du césium sur un matériau AMP/SBA-15 6 % m/m avec utilisation d’un débit ... 72
Tableau 16 : Résultats du profil d’élution d’un matériau AMP/SBA-15 6 % m/m à différents rapports Cs/AMP et concentrations de NH4NO3 après 40 ml d’éluant (n=3) ... 76
Tableau 17 : Résultats de l’adsorption de césium et de baryum dans deux milieux naturels simulés pour un matériau AMP/SBA-15 6 % m/m ... 77
Liste des abréviations et symboles
AMP Molybdophosphate d'ammonium
AMP/SBA-15 SBA-15 imprégnée de molybdophosphate d'ammonium
BET Brunauer Emmett Teller
Bq Becquerel
BuOH Butanol
C18TAB Octadecyltrimethylammonium bromide
CE Capture électronique
CI Conversion interne
CsPMo/SBA-15 SBA-15 imprégnée de phosphomolybdate de césium DFT Théorie de la fonctionnelle de la densité
DTA Analyse thermo-différentielle
EtOH Éthanol
FTIR Infrarouge à transformée de Fourier ΔG0 Énergie libre de Gibbs
Gy Gray
HPMo/SBA-15 SBA-15 imprégnée d'acide phosphomolybdique
HR-TEM Microscopie électronique en transmission à haute résolution ICP-MS Spectrométrie de masse couplée à un plasma inductif IUPAC International Union of Pure and Applied Chemistry Kd Coefficient de distribution
m/m Ratio massique
m/z Rapport masse sur charge d'un noyau atomique MAS Angle magique de rotation
MCM Mobile Composition of Matter
PEO Oxyde de polyéthylène
PPO Oxyde de polypropylène
RMN Résonance magnétique nucléaire
SBA Santa Barbara Amorphous
Sv Sievert
t1/2 Temps de demi-vie
TEOS Tetraéthyl orthosilicate
v/v Ratio volumique
XPS Spectroscopie de photoélectrons induits par rayons X Zm Isotope de masse métastable
1. Problématique
Le césium naturel, de masse atomique 133, présent habituellement à l’état de trace, se déplace beaucoup dans l’environnement, dû au fait qu’il est chimiquement très semblable au potassium, qui lui-même est un élément impliqué dans plusieurs processus chimiques. De ce fait, lorsque le radiocésium, appellation attribuée aux isotopes radioactifs du césium, est apparu dans l’environnement lors des premiers essais nucléaires, il s’est facilement intégré dans les processus environnementaux. Il existe un peu plus d’une trentaine d’isotopes radioactifs, la plupart ayant toutefois un temps de demi-vie radiologique trop court pour être intégrés aux processus environnementaux et ainsi générer une contribution dosimétrique importante. Le temps de demi-vie radiologique est la mesure du temps qu’il faut pour que la moitié de l’activité initiale disparaisse. Le tableau 1 présente les différents isotopes de césium répertoriés1. Il est à noter que CI signifie conversion interne et que CE signifie capture électronique.
Tableau 1 : Temps de demi-vie et mode de désintégration des isotopes de césium
répertoriés
Isotope Temps de
demi-vie Mode de désintégration Isotope
Temps de
demi-vie Mode de désintégration
116 3,9 s β+, CE 133 stable - 117 8 s β+, CE 134m 3,1 h CI 118 16 s CE (86 %), β+ (14 %) 134 2,2 ans β -119 38 s β+, CE 135 2,9 x 106 ans β -120 1 min β+, CE 136m 19 s CI 121 2,06 min β+, CE 136 13,2 j β -122 21 s β+, CE 137 30,2 ans β -123 6 min β+, CE 138m 2,9 min β -124 31 s β+ (92 %), CE (8 %) 138 32,2 min β- (25 %), CI 125 45 min CE (61 %), β+ (39 %) 139 9,3 min β -126 1,6 min β+ (82 %), CE (18 %) 140 1,06 min β -127 6,2 h CE (95,5 %), β+ (3,5 %) 141 24,9 s β -128 3 min β+ (61 %), CE (39 %) 142 1,69 s β -129 31 h CE 143 1,78 s β -130 30 min CE (98,4 %), β+ (1,6 %) 144 1,0 s β -131 9,7 j CE 145 0,58 s β -132 6,4 j CE (96,5 %), β- (2 %), β+ (1,5 %) 146 0,34 s β
-Ainsi, seuls trois radioisotopes sont d’intérêt d’un point de vue environnemental et sanitaire : le Cs-134, le Cs-135 et le Cs-137, avec des temps de demi-vie respectifs de 2,2 ans, 2,9 x 106 ans et 30,2 ans. Le temps de demi-vie est défini par l’équation suivante, où λ correspond à la constante de désintégration, qui est propre à chaque noyau :
[1]
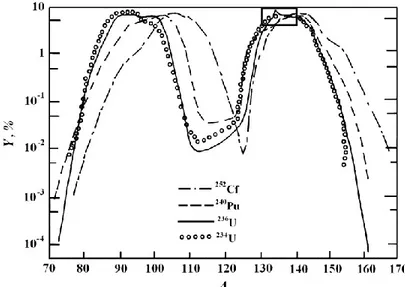
Ces radioisotopes sont essentiellement d’origine anthropique. En effet, lors de la fission binaire, soit en deux éléments différents, de masses différentes, de l’uranium, des sous-produits de désintégration sont formés dans le réacteur. Ces sous-produits sont répartis selon une distribution connue de masses qui varie selon le type de combustible utilisé et les conditions de bombardement neutroniques utilisées. Comme le montre la figure 12, il est possible de déterminer le type de combustible utilisé dans un réacteur selon les radioisotopes qui y sont produits. Selon le type de combustible, la distribution statistique des isotopes produits est différente, mais les masses des principaux radioisotopes de césium, soit 134 à 137, montrés par un encadré, correspondent au maximum de tous les types de combustibles présentés ici, d’où leur forte probabilité de production.
Figure 1 : Pourcentages relatifs des produits de désintégration du combustible nucléaire
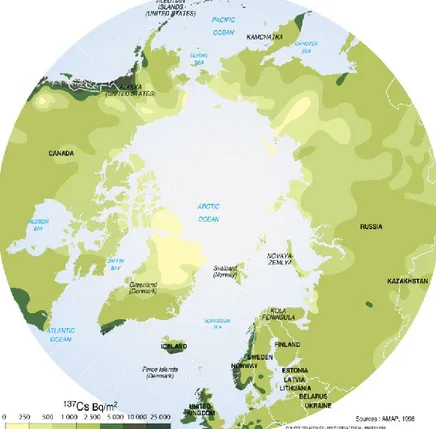
La plus grande partie de la production de radiocésium est faite dans les réacteurs nucléaires, malgré le fait qu’une petite partie de celui-ci est aussi produite dans les explosions nucléaires. Ce sont d’ailleurs ces dernières qui sont responsables de la dispersion du radiocésium dans le monde. Dans la figure 23, il est possible de voir les quantités de césium par unité de surface associées aux essais nucléaires dans le monde.
Figure 2 : Quantités en Bq/m2 de césium-137 associé aux essais nucléaires mesurées dans l’hémisphère Nord
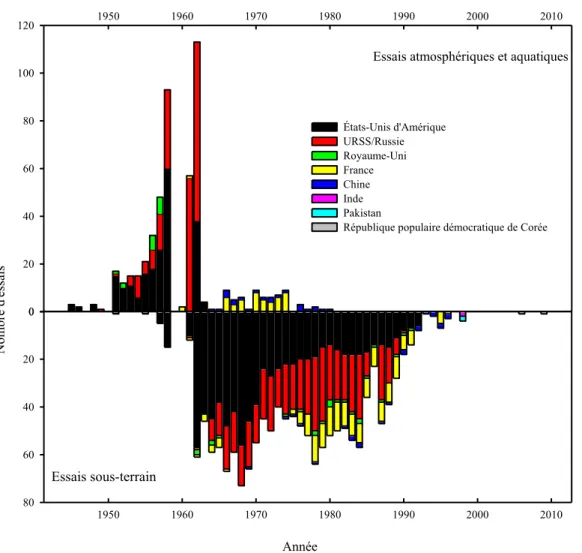
Historiquement, les essais nucléaires se sont faits en plusieurs périodes. Les premiers essais ont été faits dans l’atmosphère, entre 1945, lors des bombardements de Nagasaki et Hiroshima, et 1963, année où un moratoire a été signé entre les États-Unis d’Amérique, le Royaume-Uni et l’Union Soviétique pour arrêter les essais atmosphériques. Les essais suivants réalisés par ces pays ont donc été souterrains, et ce, à différentes profondeurs4. En France, les essais atmosphériques ont continué jusqu’en 1974 et en Chine jusqu’en 1979. Les essais souterrains ont quant à eux été poursuivis un peu partout dans le
monde, le dernier répertorié ayant été fait en Corée du Nord en 2009. La figure 35 présente les différents types d’essais nucléaires qui ont été menés selon les pays entre 1945 et 2009.
Essais atmosphériques et aquatiques
1950 1960 1970 1980 1990 2000 2010 N om br e d 'essais 0 20 40 60 80 100 120 États-Unis d'Amérique URSS/Russie Royaume-Uni France Chine Inde Pakistan
République populaire démocratique de Corée
Essais sous-terrain 1950 1960 1970 1980 1990 2000 2010 0 20 40 60 80 Année
Figure 3 : Nombre d’essais nucléaires atmosphériques et souterrains selon le pays et
l’année
Ainsi, on peut voir qu’un grand nombre d’essais nucléaires ont été faits au cours des années. Toutefois, la totalité du radiocésium dans l’environnement ne provient pas de ces essais. En effet, lors du traitement du combustible usé, il arrive que des rejets soient faits dans l’environnement, de façon intentionnelle ou non. Un cas connu de rejet fait par une d’usine de retraitement est celui de Sellafield en Angleterre. Entre 1950 et 2000, vingt et un incidents majeurs impliquant un déversement de combustible d’uranium ou de plutonium ont été relevés dans la mer d’Irlande6. Comme le combustible usé contenait encore plusieurs produits de fission lors de son retrait du réacteur, il est évident que de grandes
quantités de radiocésium ont été libérées dans l’environnement lors de ces événements. L’usine de traitement du combustible usé située à La Hague en France est un autre exemple d’installation qui rejette l’eau utilisée dans la Manche7 lors du traitement du combustible. Le césium étant difficile à séparer lors des traitements conventionnels, une grande partie de ce qui est produit est déversée chaque jour.
Deux autres événements ayant causé un grand relargage de radiocésium dans l’environnement et qui ont retenu l’attention médiatique sont les incidents arrivés dans les centrales de Tchernobyl, en 1986 en Ukraine, et de Fukushima Dai-ichi, en 2011 au Japon. Le premier a causé un relâchement d’environ 7 x 107 GBq de Cs-1374 dans l’environnement au cours des 19 jours qu’a duré l’incident. Le second événement est encore trop récent pour avoir estimé la quantité totale de césium-137 libéré lors de l’incident, il semblerait toutefois que les quantités soient comparables à celles dues à Tchernobyl8. De plus, la dispersion du radiocésium ne s’est pas faite de la même façon dans les deux cas, ce qui fait que les deux incidents ne sont pas totalement comparables. En effet, dans le cas de Tchernobyl, comme il y a eu explosion du réacteur nucléaire, la plus grande partie du radiocésium a été aéroportée, tandis que dans le cas de Fukushima, les réacteurs ont été volontairement refroidis avec de l’eau pompée et ensuite retournée dans l’océan Pacifique, ce qui a dispersé le radiocésium essentiellement dans celui-ci.
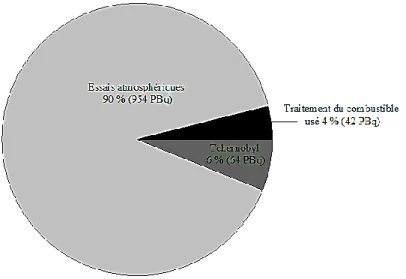
Ces incidents ont dispersé le radiocésium sur un grand territoire, mais des incidents locaux ont aussi été répertoriés, tel celui de Goiâna, au Brésil, en 1987, où une source de radiocésium sous forme de 137CsCl, utilisée à des fins médicales, a été volée et déplacée dans la ville, contaminant ainsi plusieurs centaines de personnes9. La figure 4 présente la contribution de chacun de ces modes de dispersion à la quantité totale de radiocésium présente dans l’environnement en 20074. Il est à noter que l’impact des incidents locaux tel que celui de Goîana n’a pas été considéré à cause de la trop faible quantité de radiocésium que ceux-ci ont dispersé.
Figure 4 : Principaux modes de dispersion du radiocésium et leur contribution à la quantité
totale dans l’environnement
Il a été montré à la figure 2 que la concentration en radiocésium est connue selon la région du monde. De plus, le ratio Cs-135/Cs-137 est connu selon la source de relargage. En effet, comme mentionné plus tôt, les sous-produits de désintégration varient selon les conditions présentes dans le réacteur, ce qui entraîne donc que chaque réacteur a un ratio Cs-135/Cs-137 qui lui sert de signature. D’autres ratios isotopiques sont aussi mesurés pour identifier cette signature, ils ne sont toutefois pas d’intérêt dans le cadre de ce projet. La figure 5 illustre comment il est possible de relier le rapport Cs-135/Cs-137 présent dans les sédiments de la rivière des Outaouais en aval du réacteur de Chalk River aux différentes sources qui y ont contribué10.
Figure 5 : Rapport Cs-135/Cs-137 selon la zone d’échantillonnage
Rivière Outaouais (sédiments) n=20 Bassin de sédimentation n=8 Tchernobyl n=4, SRM
Sur la figure 5, le rapport Cs-135/Cs-137 est représenté selon la distribution statistique de la zone d’échantillonnage. Ainsi, à gauche, les mesures ont été faites sur 8 échantillons pris dans le bassin de sédimentation de la centrale de Chalk River, au centre, sur 20 échantillons pris dans la rivière des Outaouais en aval de la centrale et à droite, 4 échantillons de référence pris à Tchernobyl. On peut voir que seule la colonne du centre, soit celle de la rivière des Outaouais, présente une très grande diversité de rapports Cs-135/Cs-137. Cela est dû au fait que le radiocésium des deux autres zones ne provient que d’une seule source, soit les réacteurs de Chalk River et de Tchernobyl respectivement. La grande étendue des valeurs du rapport Cs-135/Cs-137 dans le cas de la rivière des Outaouais est due au fait que le radiocésium qui y est présent provient de plusieurs sources. En effet, on peut voir que la plus grande contribution, soit dans la zone de confiance de 25 %, provient de la centrale de Chalk River, qui est située le plus près de la zone d’échantillonnage. Les valeurs inférieures sont quant à elles dues aux retombées de Tchernobyl et ne contribuent que très peu au rapport global Cs-135/Cs-137 du milieu. Finalement, les valeurs supérieures sont dues aux retombées des essais atmosphériques. Le point à l’extérieur de la zone de confiance est expliqué par le fait que la contribution en radiocésium provient de plusieurs sources différentes, qui présentent toutes une signature isotopique qui leur est propre.
Naturellement, la connaissance de ce phénomène facilite grandement l’étude de la distribution du radiocésium dans l’environnement afin de situer les dangers potentiels et de faire un suivi pour permettre de détecter les anomalies. Les connaissances ainsi acquises ont permis du même coup d’améliorer grandement la compréhension des courants aériens11, en observant la distribution et la dilution du radiocésium dans l’air après les essais atmosphériques. Toutefois, une grande partie du césium aéroporté tend à sortir de l’atmosphère, essentiellement dû à la très haute solubilité de l’ion Cs+, état d’oxydation le plus stable de cet élément.
Une fois lixivié vers le sol par les précipitations, le radiocésium tend à se complexer très rapidement à la plupart des types de sols et sédiments. Cette propriété qu’a le radiocésium de facilement intégrer les sols argileux permet de l’utiliser pour déterminer le
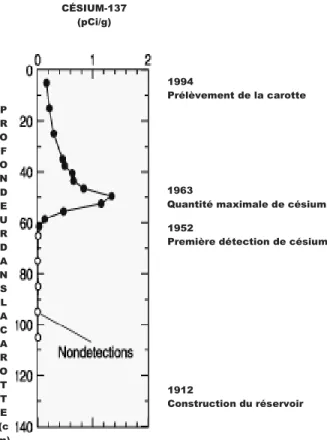
taux de sédimentation actuel d’un milieu naturel et d’ainsi déterminer l’âge des sédiments qui s’y présentent12. Pour ce faire, des carottes sont forées dans les sédiments et ensuite séparées en sections fines. En mesurant l’activité du Cs-137 dans chacune de ces sections, il est possible de déterminer celle contenant la plus grande quantité de radiocésium. Cette section correspond, dans un environnement exempt de contamination locale, à l’année 1963, soit celle suivant le maximum d’essais nucléaires, tel que montré à la figure 3. En sachant la position de la section dans la carotte de sédiments, il devient possible de déterminer le taux de sédimentation d’un milieu naturel et d’ainsi extrapoler l’âge des couches de sédiments12,13 et ainsi de fossiles et d’artefacts. La figure 614 montre un exemple d’application de ce principe.
Figure 6 : Principe de datation des sédiments par la mesure du radiocésium selon la
profondeur dans la carotte
Il est à noter que cette approche est justifiée pour des sols argileux, qui sédimentent facilement. Pour ce qui est des sols organiques, en surface, le mécanisme le plus commun pour retenir le radiocésium est l’adsorption chimique. Le cation est alors fixé par les
CÉSIUM-137 (pCi/g)
1994
Prélèvement de la carotte
1963
Quantité maximale de césium 1952
Première détection de césium
1912 Construction du réservoir P R O F O N D E U R D A N S L A C A R O T T E (c m)
substances humiques, qui rendent ainsi le radiocésium plus disponible pour les plantes. Comme mentionné plus tôt, le césium est chimiquement très semblable au potassium, ce qui fait qu’il se retrouve dans plusieurs de plantes, particulièrement les plantes inférieures15, comme les champignons, les algues et les lichens, où il y a bioaccumulation. Lors d’événements particuliers, tel que des feux de forêt, il est possible que le radiocésium soit revolatilisé du sol et des biotes vers l’atmosphère16. Toutefois, dans les conditions normales, la présence dans les plantes permet le transfert du radiocésium dans la chaîne alimentaire, où il pourrait y avoir un risque sanitaire pour l’humain.
En effet, après l’incident de Tchernobyl, la concentration en radioisotopes a été mesurée dans des aliments tels que le lait et la viande. À partir de ces analyses, des coefficients de transfert et de rétention ont été établis pour chaque type de lait et d’animal pour prévoir au final quels aliments étaient les plus dangereux pour la population humaine. De ces études est ressorti que la volaille, les œufs et le lait de chèvre contenaient le plus de radiocésium17 et qu’il fallait surveiller ces aliments plus attentivement.
La connaissance de la quantité de radiocésium présente dans un être vivant est aussi très utile en ce qui concerne l’estimation de doses qu’un animal ou bien qu’un humain aurait reçu. Les traitements subséquents à une incorporation d’un radioisotope nocif dans le corps est coûteux et peuvent être dangereux pour la personne18, ce qui fait que ce type de traitement est seulement utilisé lorsqu’une dose assez importante a été mesurée. Toutefois, pour estimer ce niveau, il faut tenir compte du temps de demi-vie réel du radioisotope dans le corps. En effet, en plus du temps de demi-vie du radiocésium, il faut savoir le temps de résidence de celui-ci dans le corps humain. Ce dernier dépend des différents mécanismes d’assimilation du radioisotope par le corps et dépend de la nature chimique de l’élément, de l’âge et de l’état de santé de l’individu ainsi que du mode d’exposition de l’organisme, soit par ingestion, inhalation ou contact cutané. Ainsi, un suivi immédiat après l’accident de Tchernobyl a permis de quantifier la quantité relative de radiocésium assimilée par l’organisme chez des personnes de tous âges et un suivi historique après l’incident de Goiâna a permis de situer le temps de demi-vie biologique entre 2 et 110 jours, selon l’âge de la personne et le mode d’assimilation dans le corps19. Ces études ont permis une
compréhension plus approfondie du comportement du radiocésium dans le corps, ce qui facilite donc beaucoup la gestion des traitements lors d’incidents impliquant l’épanchement de radiocésium.
1.1 Techniques d’analyse du césium
Il a été vu à la section précédente que la connaissance de la concentration des principaux radioisotopes de césium est importante, tant au point de vue scientifique, en météorologie ou en géochimie, qu’au point de vue médical et sanitaire. Il existe trois techniques principalement utilisées pour mesurer le radiocésium.
Tout d’abord, il faut savoir quel type d’émetteur est chaque isotope pour faciliter son analyse. Il existe trois types de rayonnements principaux : alpha (α), bêta (β), et gamma (γ). Le rayonnement α est en fait une particule constituée de deux neutrons et de deux protons, l’équivalent d’un noyau d’hélium chargé 2+. Cette particule est très énergétique, mais du fait de sa taille et de sa charge, elle est facilement arrêtée. Le rayonnement β, lui, est constitué d’un électron, β-, ou d’un positron, β+. Il est moins énergétique que la particule α, mais comme il possède une masse très faible, il pénètre plus facilement dans la matière. Finalement, le rayonnement γ est une onde électromagnétique comparable aux rayons X (photon gamma).
Le césium-134 et le césium-137 sont tous les deux des émetteurs β- et γ; le césium-135, quant à lui, est un émetteur β- pur, tel que vu au tableau 1. Ainsi, quand il s’agit de faire une analyse isotopique peu de temps après un événement, il est facile d’utiliser la spectrométrie gamma pour doser les radiocésium. Le principe de la spectrométrie γ est simple. Il s’agit de la formation d’un courant électrique dans un détecteur semi-conducteur, généralement du germanium de haute pureté, à partir de la radiation que celui-ci reçoit. Pour éviter le plus d’interférences possible, la chambre de comptage est isolée au plomb et le détecteur est refroidi à l’azote liquide. Ce dernier point sert à éviter qu’il y ait conduction à cause de la chaleur ambiante.
La scintillation liquide est utilisée pour des radiations de faible énergie, elle n’est donc pas utile pour le rayonnement γ. Pour cela, il s’agit d’immerger l’échantillon dans un mélange de solvant, très souvent du toluène à cause de son grand nuage électronique π, et de scintillant. Un scintillant est une molécule, qui possède généralement des cycles aromatiques conjugués, et qui phosphoresce ou fluoresce lorsqu’elle est excitée. Ainsi, lorsque l’échantillon est couvert de cette solution, les particules ionisantes qu’il émet interagissent avec le solvant qui capte cette énergie et la transmet ensuite au scintillant. Le scintillant émet alors un rayonnement lumineux que pourra mesurer le détecteur. Cette technique serait applicable dans le cas du radiocésium, toutefois, elle est limitée dans le cas du Cs-135, qui possède un temps de demi-vie très long. De plus, la scintillation liquide ne fait pas de distinction entre les différents rayonnements et les énergies de ceux-ci, rendant cette technique plus utile pour la mesure d’une dose totale plutôt que pour différencier les isotopes.
Comme les temps de demi-vie des isotopes 134 et 137 sont relativement courts, le comptage se fait facilement. Par contre, lors d’événements plus lointains, comme c’est le cas avec Tchernobyl, il n’est plus possible de détecter le Cs-134, puisque celui-ci aura disparu de l’environnement. En effet, lorsque le temps écoulé depuis l’événement correspond à dix fois le temps de demi-vie du radionucléide d’intérêt, celui-ci n’est plus considéré comme présent dans le milieu. Ainsi, il faut plutôt se tourner vers la spectrométrie de masse inorganique pour analyser l’isotope de masse 135. Le fait que sa demi-vie soit très longue (2,9 x 106 ans) permet de facilement le doser par spectrométrie de masse inorganique (ICP-MS). Il en est de même pour le Cs-137. Néanmoins, l’application de cette technique aux isotopes de masses 135 et 137 a ses désavantages. En effet, comme la chaîne de désintégration de ces isotopes finit avec un atome de baryum de masse stable dans les deux cas, cela crée une interférence isobarique lors de la mesure. La figure 720 présente la chaîne de désintégration du Cs-137. On peut y voir que le Cs-137 se désintègre selon deux possibilités différentes, toutes deux avec émission de β-, mais d’énergies différentes. La première est une désintégration β- suivie d’une émission γ ou d’une conversion interne, la seconde est une émission β- pure. Les pourcentages indiqués dans la
Figure 7 : Chaîne de désintégration du césium-137
La formation de ces interférences isobariques nécessite donc une séparation préliminaire à l’analyse par spectrométrie de masse inorganique. De plus, comme le baryum est environ 100 fois plus abondant dans l’environnement que le césium, cela rajoute une difficulté de plus à l’analyse de la masse 137 en spectrométrie de masse. Cette séparation sert aussi à préconcentrer le radiocésium. En effet, comme ces radioisotopes sont généralement présents en très faibles quantités dans l’environnement ou dans les êtres vivants, la préconcentration des analytes permet d’améliorer les limites de détection et de quantification de la méthode de dix à mille fois.
1.2 Techniques de séparation du césium
Différentes techniques ont été mises au point pour la séparation du césium des différents interférents possibles, que ce soit lors de l’analyse par émission γ ou par spectrométrie de masse inorganique.
1.2.1 Chromatographie ionique
La chromatographe ionique est une technique qui utilise le principe d’échange d’ions, généralement avec des ligands organiques, pour capter et retenir des ions
préférentiellement à d’autres. Pour cela, il est possible d’utiliser des résines échangeuses d’anions ou de cations, et de modifier les charges des ions concernés pour les retenir ou bien les éluer. Dionex, une des compagnies qui fabriquent des résines commerciales, utilise souvent comme support inerte des billes de polystyrène ou de polyvinylbenzène21. De même, la compagnie Dowex utilise des amines quaternaires ainsi que des sulfonates comme agents complexants sur ces mêmes types de supports.
Alternativement, plusieurs groupes de recherche22,23 se sont aussi orientés vers la complexion du césium et autres alcalins et alcalino-terreux par les éthers-couronne et calixarènes. Les éthers-couronnes sont des macrocycles formés d’oxyéthylène (CH2-CH2
-O), tandis que les calixarènes sont des macromolécules constituées de cycles aromatiques reliés entre eux par des chaînes de carbone. Une structure de chaque type est présentée à la figure 8. La différence majeure entre ces deux types de composés est la structure bidimensionnelle des éthers-couronne et la structure tridimensionnelle des calixarènes.
Figure 8 : Exemples d'éther-couronne (gauche) et de calixarène (droite)
Il est toutefois de plus en plus commun d’associer les deux types de structures pour capter et retenir les ions plus facilement. En effet, comme il est facile de modifier la taille ainsi que la polarité de ces « cages », celles-ci deviennent un outil de choix pour la complexion ionique dans plusieurs milieux.
1.2.2 Ferrocyanates de métaux alcalins
Les ferrocyanates, de structure générale A2nM2-nFeCN·xH2O, où A est un cation
monovalent (ex : K+, Cs+, NH4+) et M un métal divalent ou un cation divalent comme
TiO2+, 24 sont des solides utilisés pour l’adsorption du césium. Deux réactions sont utilisées pour cela, la première étant un échange d’ion, soit généralement le potassium pour le césium, la deuxième, plus complexe, inclut la réduction du fer, normalement trivalent, pour l’amener à un état divalent, permettant ainsi d’accepter une nouvelle charge positive dans le complexe. Les ferrocyanates de potassium et de nickel sont très communs25,26,27, mais le cuivre28,29 et le cobalt30 sont aussi très utilisés à cause de leur similitude chimique avec le nickel. Habituellement, les ferrocyanates sont stables dans les milieux neutres autant que dans les milieux basiques30.
Plusieurs groupes ont essayé de synthétiser les ferrocyanates sur un support pour améliorer la stabilité et la réutilisabilité du système. Ainsi, Rao29 a synthétisé du CuFeCN dans des cubes de polyuréthane qu’il utilisait en les agitant dans la solution de césium. De même, Konecny a synthétisé un matériau dans un gel de silice31. Sangvanich, quant à lui, a synthétisé un matériau de silice mésoporeuse de type MCM-41 sur lequel il a greffé de l’éthylène diamine pour la faire complexer avec du cuivre pour mener à la formation d’un complexe CuFeCN32.
Ce genre de matériaux a habituellement une bonne stabilité chimique et thermique, ainsi qu’une bonne résistance aux radiations. Toutefois, la sélectivité n’est pas très bonne envers plusieurs cations monovalents, spécialement envers le thallium32 et le potassium27.
1.2.3 Molybdophosphate d’ammonium
Le molybdophosphate d’ammonium, de formule (NH4)3PMo12O40, est un sel faisant
partie d’un regroupement cristallin appelé structure de Keggin. La structure de Keggin est constituée d’un tétraèdre PO43- entouré de douze octaèdres MoO6 dont deux oxygènes sont
partagés pour relier les octaèdres entre eux. Ces douze unités sont arrangées en quatre groupes pour donner la structure présentée à la figure 933.
Figure 9 : Structure de Keggin
Comme l’unité de Keggin est très grande et que les interstices sont nombreux, le transport de cations à l’intérieur de celle-ci devient aisé et rapide34. Il a été montré que cela vient du fait que les anions de Keggin sont isomorphes35, c’est-à-dire que leur structure cristalline ne change pas lorsqu’il y a échange de cations. De plus, il est connu que le molybdophosphate d’ammonium (AMP) montre une très grande sélectivité envers certains cations plutôt qu’envers d’autres. Le tableau 236 présente les énergies libres de Gibbs lors de l’échange de l’ammonium pour un autre cation monovalent.
Tableau 2 : Énergies libres de Gibbs de différents échanges de cations pour l'AMP Cation ΔG° (J/mol) K+ 3 140 ± 400 Rb+ -5 690 ± 630 Cs+ -11 880 ± 1 250 Tl+ -11 630 ± 1 250 Ag+ 3 550 ± 400
À partir de ce tableau, on peut voir que certains échanges sont largement favorisés par rapport à d’autres; c’est le cas du césium et du thallium. Il n’est pas tout à fait clair pourquoi le thallium agit de façon similaire au césium, mais comme vu à la section précédente, le même problème se voit avec les ferrocyanates de métaux alcalins. Malgré l’échange favorisé de l’ammonium pour le césium, il est aussi possible de revenir facilement vers le composé de départ, soit l’AMP, en utilisant un excès d’ammonium en
solution. En effet, Coetzee a déterminé que malgré l’adsorption d’autres cations, comme le sodium et le potassium, l’AMP reste sélectif au césium37.
En plus de posséder une remarquable capacité d’échange d’ions, l’AMP est très stable sur une large gamme de pH, ce qui permet de l’utiliser dans un milieu très acide ou neutre30. De plus, il est très résistant aux radiations du fait de la nature ionique de ses liaisons. Rao38 et Narasimharao39 ont tous deux prouvé que l’AMP ne se dégradait pas lorsque irradié avec des électrons et Oldham40 a montré que le molybdophosphate d’ammonium résiste aussi au bombardement neutronique. D’autre part, l’AMP est aussi très stable à haute température, jusqu’à environ 410 °C.
1.2.3.1 Molybdophosphate d’ammonium supporté
Malgré sa très grande efficacité pour l’adsorption du césium et sa grande stabilité chimique et radiologique, le molybdophosphate d’ammonium est très peu utilisé à grande échelle puisqu’il forme de très petits cristaux qui favorisent de faibles débits et un blocage rapide dans les colonnes. Pour cela, plusieurs groupes se sont penchés sur différents types de supports inertes qui pourraient convenir à l’AMP et former un matériau stable et utilisable à grande échelle.
1.2.3.1.1 Supports organiques
Le type de support organique le plus commun est celui des polymères, particulièrement le polyacrylonitrile41,42. Celui-ci est choisi à cause de sa stabilité dans une large gamme de pH. Cependant, il ne présente pas de lien chimique avec le molybdophosphate d’ammonium, ce qui le rend moins stable à long terme. De plus, dans les deux cas, l’AMP a été lavé pour récupérer le césium, ce qui rend impossible la réutilisation du matériau, malgré le retour facile du molybdophosphate de césium vers le molybdophosphate d’ammonium.
D’autre part, Stejskal43 a essayé différents types de polymères : du polystyrène, des méthacrylate, de l’époxy ainsi que des polymères vinylés. Il en est venu à la conclusion que les systèmes semblaient stables, mais qu’ils se détérioraient au-delà de 40 °C.
Comme autre système, Mimura44 a présenté un matériau de molybdophosphate d’ammonium immobilisé dans de l’alginate de calcium. Le système est efficace pour de petites quantités de césium à extraire malgré le fait que l’étude ne présente pas la stabilité à long terme du matériau ni sa résistance aux radiations.
Finalement, Epov45 a essayé des billes de téflon comme support inerte, sur lesquelles l’AMP a été déposé. Le système semble stable, mais l’élution avec une solution contenant de l’ammonium n’a pas été tentée, le molybdophosphate a plutôt été dissout pour libérer le césium.
1.2.3.1.2 Supports inorganiques
Différents supports inorganiques ont aussi été testés quant à leur utilité pour un système avec l’AMP. L’amiante a été utilisé à plusieurs reprises, puisque, comme l’AMP, il est très stable en milieu acide. Coetzee37 l’a utilisé pour montrer qu’il est possible de créer une méthode efficace pour la séparation du césium dans un milieu acide. Il n’a toutefois pas essayé d’éluer le césium. Van R. Smit46 a quant à lui tenté l’élution avec une solution saturée de nitrate d’ammonium, qui semblait fonctionner correctement. Cependant, son système n’a pas été testé dans des conditions vérifiant sa stabilité aux radiations. Finalement, Dutta-Roy47 a essayé l’élution du césium-137 dans un système similaire. Celle-ci semble d’ailleurs efficace, mais seulement pour de petites quantités, soit 35 690 Bq, ce qui représente environ 10-5 mg de Cs-137 pour 3 g de molybdophosphate d’ammonium.
Le gel de silice a aussi été utilisé à des fins d’immobilisation de l’AMP, puisqu’en plus d’offrir une grande stabilité thermique, chimique et radiative, elle présente une bonne porosité qui permet d’immobiliser efficacement une grande quantité d’AMP, sans toutefois nuire à une bonne circulation d’un liquide dans le matériau. Ainsi, Van So48 a synthétisé un
matériau d’AMP sur gel de silice pour vérifier sa stabilité thermique. De son côté, Stejskal49 a synthétisé un système du même genre et y a testé l’adsorption et l’élution du césium. Il n’a cependant pas vérifié la stabilité thermique ou radiative du matériau.
Un autre protocole qui a conduit à un matériau stable d’AMP dans un support inorganique est celui de Tranter50. Ce dernier utilise la co-condensation pour former des billes poreuses d’aluminosilicate intégrant dans leur structure l’AMP. Le système est stable aux radiations ainsi que dans des milieux acides et efficace pour l’adsorption d’une grande quantité de césium, mais l’adsorption se fait lentement et l’élution n’a pas été tentée.
1.3 Matériaux poreux et ions de Keggin
Le point commun des deux derniers types de matériaux, soit le gel de silice et les billes d’aluminosilicate, est leur grande porosité. La porosité dans un matériau lui confère une grande surface disponible dans les pores, permettant ainsi d’y déposer ou d’y attacher une grande quantité de réactif, mais aussi de pouvoir créer un débit dans les pores de ce matériau grâce à leur grand volume poreux. Pour cette raison, les matériaux poreux, spécialement les matériaux mésoporeux sont très utilisés pour y imprégner des ions de Keggin, qui servent souvent de catalyseurs pour oxyder sélectivement les alcènes. Liu51 a synthétisé des matériaux d’acide phosphotungstique sur de la silice mésoporeuse SBA-15 pour l’isopropylation du naphtalène; Predoeva52, des acides phosphomolybdovanadiques sur des oxydes de titane mésoporeux pour l’oxydation sélective du chlorobenzène; Bordoloi53, des acides phosphomolybdovanadiques sur des silices mésoporeuses hexagonales (HMS) pour l’oxydation sélective du propylène; et Benadji54, des acides phosphomolybdo-vanadiques sur des silices mésoporeuses SBA-15, MCM-41 et MCM-48 pour l’oxydation sélective de l’anthracène.
Ce même genre de matériaux peut aussi être utilisé comme catalyseurs pour l’alkylation sélective des phénols. Ainsi, Wang55 a synthétisé des matériaux Cs3PW12O40
sur silice mésoporeuse MCM-41; Kozhevnikov56,57 et Kumar58 des matériaux d’acide phosphotungstique sur des MCM-41 pour l’alkylation des phénols.
Pour d’autres utilisations, Pizzio59 a synthétisé des acides phosphomolybdique et phosphotungstique sur de l’oxyde de zirconium mésoporeux; Dias60, du phosphotungstate de césium sur MCM-41; et Kim61,62, de l’acide phosphomolybdique lié à de l’aminopropyltriethoxysilane, lui-même greffé sur de la silice MCF pour la conversion de l’éthanol.
D’un point de vue plus théorique, Jin63 a synthétisé des matériaux d’acide germanotungstovanadique sur silice mésoporeuse SBA-15 pour en étudier la stabilité thermique, et Kasztelan64,65, des matériaux d’acide phosphomolybdique sur des silices mésoporeuses pour étudier leur interaction avec le césium.
1.4 Silices mésoporeuses
Les silices mésoporeuses sont nommées ainsi à cause de la taille de leurs pores. L’IUPAC a classé les matériaux poreux en trois catégories : les microporeux, dont les pores sont inférieurs à 2 nm de diamètre, les mésoporeux, avec des pores compris entre 2 et 50 nm, et les macroporeux, dont le diamètre des pores est supérieur à 50 nm66. Elles sont caractérisées par une porosité ordonnée, une grande surface spécifique et un grand volume poreux. De plus, elles sont facilement modulables dans ces paramètres et facilement fonctionnalisables67.
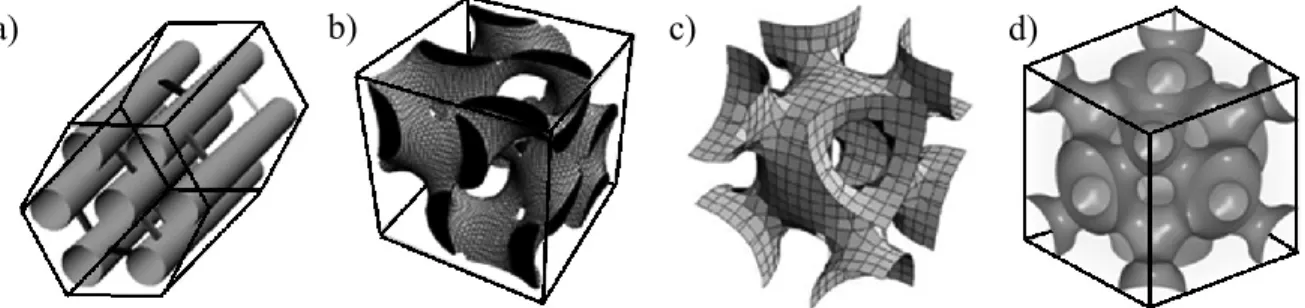
Les silices mésoporeuses sont divisées en plusieurs familles selon le type de géométrie de pores et la méthode de synthèse. Ainsi, il existe plusieurs géométries différentes, dont quatre sont présentées dans la figure 1068. De gauche à droite, la structure bidimensionnelle hexagonale (p6mm), la structure tridimensionnelle bicontinue (Ia d), la structure tridimensionnelle ouverte cubique centrée (Im m) et la structure tridimensionnelle cubique à face centrée (Fm m). Un exemple typique de chacune serait la SBA-1569 pour le réseau bidimensionnel hexagonal, la KIT-670 pour la tridimensionnelle continue, la SBA-1671 pour la structure ouverte cubique centrée et la KIT-5 pour la structure face centrée72.
Figure 10 : Exemples de structures des silices mésoporeuses : a) p6mm, b) Ia d, c) Im m,
d) Fm m
Ces structures peuvent être obtenues en modifiant l’agent tensioactif (servant d’espèce porogène), le précurseur de silice et le pH du milieu. Pour modifier l’épaisseur de paroi, la taille des pores et l’interconnectivité des pores dans le cas des p6mm, il est possible de varier le temps et la température durant la synthèse. Malgré les différences dans les paramètres fins de la synthèse, le principe à la base du protocole reste le même. Celui-ci est présenté dans la figure 1173. Les explications sont adaptées à la synthèse de la SBA-15 puisque celle-ci sera d’importance dans ce projet.
Figure 11 : Procédé de synthèse des silices mésoporeuses bidimensionnelles hexagonales
(ex. SBA-15)
Lors de la première étape, l’agent structurant, ou tensioactif, dans le cas de la SBA-15, le Pluronic P123, un copolymère-bloc PEO20-PPO70-PEO20 est dissout dans une
solution aqueuse acidifiée avec de l’acide chlorhydrique. La concentration de l’agent structurant est alors très importante puisque ce sont les interactions entre le solvant et l’agent structurant, ainsi que les interactions entre les molécules de l’agent tensioactif qui va déterminer s’il y aura formation de micelles aussi bien que la forme de celles-ci. Ainsi, une faible concentration entraînera la mise en solution des molécules, une concentration un peu plus élevée assurera la formation de micelles sphériques, et à plus forte concentration, les micelles auront plutôt une forme cylindrique et auront tendance à s’agréger pour former le réseau hexagonal, allant jusqu’à former des cristaux liquides à très haute concentration. Comme les molécules du copolymère-bloc sont amphiphiles, seule la partie polaire aura tendance à rester en contact avec la solution aqueuse. Plus la concentration de l’agent tensioactif augmentera, plus l’entropie du milieu augmentera, favorisant ainsi la formation d’une structure qui permettra de diminuer celle-ci. C’est au cours de ce processus que le précurseur de silice, soit le tetraéthylorthosilicate (TEOS), est ajouté. L’acidité du milieu favorise son hydrolyse et sa condensation autour des micelles. Ce procédé est appelé auto-assemblage coopératif74,75,76.
Par la suite, une étape de consolidation est faite, où il s’agit de faire un traitement hydrothermique, de durée et de température variables, afin de condenser le réseau de silice pour augmenter la taille et l’interconnectivité des pores77 de même que la résistance hydrothermique du matériau final. Finalement, l’agent tensioactif est éliminé, très souvent par calcination à 550 °C. Il est possible toutefois d’utiliser une extraction au solvant ou un traitement aux micro-ondes76.
1.5 Objectifs du projet
En combinant les certitudes qu’il est possible de synthétiser des matériaux contenant des unités de Keggin sur des silices mésoporeuses et celle que le molybdophosphate d’ammonium possède des qualités de stabilité et d’efficacité quant à l’adsorption et à l’élution de césium, il a été supposé qu’il était possible de synthétiser un matériau qui présenterait les avantages des silices mésoporeuses et de l’AMP. Ce matériau serait stable
dans des conditions d’acidité, de radioactivité, de température et de pression élevées, présenterait une bonne surface spécifique pour l’échange de cations, ne causerait pas de blocage lorsqu’utilisé en colonnes et serait réutilisable un nombre indéfini de fois, diminuant ainsi les déchets liés à son utilisation.
Pour cela, il faut choisir un support de silice mésoporeuse qui présente les caractéristiques voulues. En effet, il serait préférable d’avoir des pores très grands pour permettre un bon passage du liquide lors de l’utilisation, mais des parois épaisses pour ne pas diminuer la stabilité thermique. La silice mésoporeuse SBA-15 serait donc idéale puisqu’elle présente ces caractéristiques. Il s’agit toutefois ici de l’élaboration d’un modèle applicable par la suite à d’autres types de supports.
Ainsi, l’objectif principal de ce projet est de synthétiser un matériau de molybdophosphate d’ammonium immobilisé sur une silice mésoporeuse SBA-15 pour l’adsorption et l’élution efficaces du radiocésium, et ce dans le but de réutiliser le matériau.
2. Techniques de caractérisation
2.1 Physisorption de gaz
La physisorption de gaz est une technique utilisée pour qualifier et quantifier la porosité de matériaux poreux. La méthode est basée sur le principe de l’adsorption physique d’un gaz sur un solide. Différents modèles mathématiques ont été développés pour analyser les forces qui sont en jeu lors de ce procédé, soit les forces de van der Waals et les forces de répulsion intermoléculaires78. Une analyse se passe ainsi : premièrement, il faut évacuer sous vide et avec un léger chauffage le matériau d’intérêt pour enlever tout solvant ou eau adsorbée et ainsi avoir la masse réelle de l’échantillon. Deuxièmement, le gaz qui joue le rôle de de l’adsorbat, généralement l’azote, est injecté peu à peu dans l’échantillon refroidi à la température de condensation du gaz, dans le cas de l’azote, à -196 °C, et une mesure du volume injecté est prise à diverses pressions après un temps laissé pour l’obtention de l’équilibre. Cela produit donc un graphique du volume de gaz adsorbé en fonction de la pression relative P/P0, à température constante (isotherme), où P0
correspond à la pression de saturation du gaz. L’IUPAC (International Union of Pure and Applied Chemistry) a classé les isothermes les plus communes en six catégories, qui sont présentées à la figure 1279.
Figure 12 : Classification des isothermes de physisorption selon l’IUPAC
Dans le cas de ce projet, seul l’isotherme de type IV sera montré, puisqu’il correspond à l’adsorption dans des matériaux ayant des mésopores relativement grands, comme dans le cas de la SBA-15.
De plus, comme il est montré à la figure précédente, certains types d’isothermes présentent une hystérèse. Pour aider à la distinction des différentes isothermes, ces hystérèses ont-elles aussi été classées en différents groupes. La figure 1379 présente cette classification.
Figure 13 : Classification des hystérèses selon l'IUPAC
Comme ce projet ne présentera que des matériaux SBA-15 ou des matériaux de SBA-15 imprégnée, seul le type H1 sera vu. Celui-ci correspond à un réseau poreux organisé avec des pores de forme cylindrique.
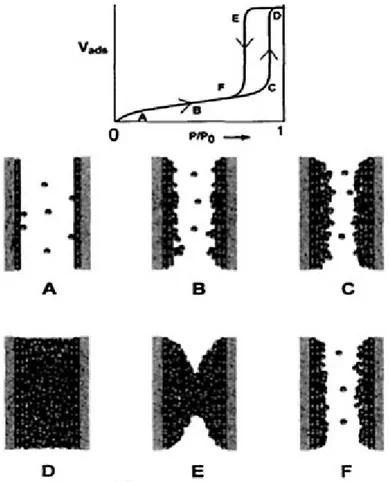
Chaque isotherme peut être décomposée en plusieurs parties, qui correspondent à une phase de l’adsorption ou de la désorption de l’azote dans le matériau. La figure 1480 présente une explication de ce procédé.
Figure 14 : Isotherme d’une SBA-15 expliquée par le processus d'adsorption des molécules
d'azote dans un pore
Le processus commence en A avec la formation d’une monocouche de molécules d’azote sur la surface interne du pore. C’est la région où il est possible de déterminer la microporosité et la surface spécifique du matériau. En B, l’adsorption de gaz commence à former une multicouche de molécules. La montée rapide du volume adsorbé en C indique qu’il y a condensation capillaire, due au fait que les forces intermoléculaires entre les molécules d’azote sont rendues plus importantes que les forces d’attraction qui existent entre les parois et l’adsorbat. Le plateau atteint en D indique que le pore est saturé en azote. C’est à cette pression relative que le volume poreux total est généralement calculé. Ensuite, lors de la désorption, l’hystérèse en E est due aux mêmes forces qui agissaient lors de l’adsorption et qui causaient la montée rapide du volume adsorbé. Dans le cas de pores cylindriques quasi-parfaits, la formation de l’hystérèse est habituellement due à des différences thermodynamiques au niveau du comportement du fluide confiné dans les nanopores, ce qui fait que les processus d’adsorption et de désorption ne se produisent pas
de manière réversible. La condensation capillaire est retardée, car le gaz étant adsorbé sous forme de film, il reste métastable jusqu'à nucléation d’une interface gaz-liquide, alors que la désorption se produit à la pression d’équilibre liquide-gaz. De plus, la présence de défauts dans les structures poreuses cylindriques, par exemple des constrictions, peut aussi retarder la désorption78. Finalement, en F, les pores se vident jusqu’à ce qu’il ne reste qu’une monocouche, qui elle aussi disparaît lors de la diminution de la pression.
Habituellement, des modèles mathématiques sont utilisés pour calculer la surface spécifique et la taille et le volume poreux. Pour la surface spécifique, comme mentionné plus haut, il s’agit d’utiliser le volume injecté lors de la formation de la monocouche d’azote dans les basses pressions relatives, soit entre 0,05 < P/P0 < 0,2, auquel on applique
la méthode BET, soit Brunauer Emmett Teller. En simplifiant, le volume adsorbé de gaz dans une monocouche est converti en aire spécifique. Le volume poreux est mesuré à P/P0 = 0,95, ce qui correspondant au plateau atteint à la fin de l’adsorption, donc au
remplissage complet des pores. Finalement, il est possible d’utiliser la dérivée des volumes poreux cumulés, associés à des méthodes d’analyses théoriques basées sur la DFT, pour obtenir une distribution de tailles de pores assez réaliste81.
2.2 Microscopie électronique et analyse dispersive en énergie
La microscopie électronique est un outil qui sert à voir la matière à un niveau atomique. En effet, il est possible avec cette technique de voir des objets qui ne font que quelques nanomètres, ce qui ne correspond qu’à quelques centaines d’atomes d’épais. La technique est basée sur la réaction qu’a la matière lorsqu’elle est frappée par des électrons de haute énergie. Pour cela, une source d’électrons, souvent un filament de tungstène, est installée dans un milieu sous haut vide. Les électrons qui sont émis sont ensuite concentrés et redirigés par des lentilles magnétiques vers l’échantillon. Celui-ci va les diffracter selon un certain angle et avec plus ou moins de perte d’énergie ou bien absorber l’énergie et émettre des rayons X82. Dans les deux cas, un détecteur est placé dans la chambre d’analyse pour les retransmettre à l’ordinateur qui les transforme en images.
2.3 Diffraction des rayons X à bas et grands angles
Lors de la diffraction des rayons X, l’échantillon est bombardé par une source de rayons X possédant une énergie variant entre 3 et 8 keV. Comme les longueurs d’onde utilisées pour ce type d’analyse sont du même ordre de grandeur que les liaisons atomiques, soit dans l’ordre de la dizaine d’Angströms, il y a possibilité de diffraction des rayons X. Pour que ce phénomène soit quantifiable, il faut toutefois que les atomes soient arrangés d’une manière ordonnée, donc cristalline. Cette diffraction est présentée à la figure 1583.
Figure 15 : Diffraction des rayons X dans un cristal
La compréhension de la diffraction dans un cristal a mené à la création de la loi de Bragg, qui est donnée par l’équation suivante :
[2]
où n provient des plans présents dans le cristal et est propre à chaque structure cristalline, λ est la longueur d’onde de la source des rayons X, d, la distance interplanaire et θ, l’angle de diffraction mesuré84. Comme d dérive des paramètres de maille de la structure cristalline, il est facile par la suite de revenir à ceux-ci.
2.4 Résonance magnétique nucléaire du phosphore-31 à l’état solide
Pour comprendre la résonance magnétique nucléaire, il est important de savoir que les noyaux atomiques ont un spin, qui est de ½ pour le phosphore-31. Ce spin nucléaire provoque un moment angulaire qui peut être modifiable avec l’application d’un champ magnétique. La figure 16 montre comment ce moment angulaire peut être modifié.
Figure 16 : Mouvement de précession d'un noyau en RMN
L’application d’un champ magnétique B0 sur le moment angulaire µ provoque
l’alignement de ce dernier sur B0. Lorsque le champ magnétique est arrêté, le noyau prend
un certain temps avant de revenir à son moment angulaire initial, il s’agit du temps de relaxation. Ce temps varie selon l’environnement chimique du noyau, ce qui signifie qu’il est affecté par les autres noyaux et électrons présents autour de lui. À partir de ce temps, un signal est calculé, auquel on applique une transformée de Fourier pour le transformer en déplacement chimique.
En RMN du solide, la technique diffère quelque peu. En effet, comme les mouvements des molécules de l’échantillon sont restreints par l’état solide de celui-ci, l’anisotropie, soit la dépendance à la position, et le couplage dipolaire, sont présents. Ces phénomènes sont habituellement nuls en RMN à l’état liquide puisque les mouvements moléculaires finissent par se compenser mutuellement. Pour contrer ces phénomènes, il faut faire tourner l’échantillon à un angle magique (Magic Spinning Angle) de 54,7 ° à une vitesse plus grande que 2 kHz85.