Calcific aortic valve disease is, by far, the most prevalent form of aortic stenosis (AS) worldwide. In the develop-ing world, AS may also be caused by rheumatic heart disease. Calcific aortic valve disease is characterized by fibro-calcific remodelling of the valve leaflets. In the first phase of the disease, termed aortic sclerosis, the valve becomes thickened and mildly calcified but these changes do not cause any obstruction to blood flow. Over the years, the disease evolves to severe valve calcification with impaired leaflet motion and vast blood flow obstruction, which are hallmarks of calcific AS1
(TABLE 1). In developed countries, AS is the third-most common cardiovascular disease after coronary artery disease and systemic arterial hypertension2. Over the
past five decades, the management of calcific AS has changed dramatically. Doppler echocardiography has replaced cardiac catheterization as the method of choice for the diagnosis and follow-up of AS, and transcatheter valve therapy has emerged as an alternative to surgery for aortic valve replacement (AVR). However, no pharma-cotherapy has proved to reduce either the progression of valve stenosis or the resulting adverse effects on left ventricular function and patient outcomes. Hence, surgi-cal or transcatheter AVR are the only effective treatment
options for severe AS3,4. Overall, this disease is directly
responsible for approximately 85,000 AVRs and 15,000 deaths per year in North America2. In this Primer, we
discuss the epidemiology, mechanisms, diagnosis and management of calcific AS, and highlight how the intro-duction of transcatheter-based valve replacement has transformed patient outcomes.
Epidemiology
Calcific AS is the consequence of progressive fibro- calcific remodelling occurring on an initially normal (tricuspid) aortic valve or a congenitally abnormal (bicuspid) aortic valve. Although the prevalence of bicuspid aortic valve is only 0.5–1.0% in children, it accounts for nearly half of aortic valves that are sur-gically removed because of calcific AS5. During their
lifetime, most individuals with a bicuspid aortic valve develop some kind of aortic valve pathology, the most common being AS5–8. Furthermore, patients with
bicus-pid valve develop calcific AS one or two decades earlier than those with a tricuspid valve.
Aortic sclerosis, which is the preclinical phase of calcific aortic valve disease, is defined as focal areas of valve calcification and leaflet thickening without
Correspondence to P.P. Québec Heart and Lung Institute, Department of Medicine, Laval University, 2725 Chemin Sainte-Foy, Québec City, Québec G1V 4G5, Canada.
philippe.pibarot@ med.ulaval.ca
Article number: 16006 doi:10.1038/nrdp.2016.6 Published online 3 Mar 2016
Calcific aortic stenosis
Brian R. Lindman
1, Marie-Annick Clavel
2, Patrick Mathieu
2, Bernard Iung
3,4,
Patrizio Lancellotti
5,6, Catherine M. Otto
7and Philippe Pibarot
2Abstract | Calcific aortic stenosis (AS) is the most prevalent heart valve disorder in developed
countries. It is characterized by progressive fibro-calcific remodelling and thickening of the aortic
valve leaflets that, over years, evolve to cause severe obstruction to cardiac outflow.
In developed
countries, AS is the third-most frequent cardiovascular disease after coronary artery disease and
systemic arterial hypertension, with a prevalence of 0.4% in the general population and 1.7% in the
population >65 years old. Congenital abnormality (bicuspid valve) and older age are powerful risk
factors for calcific AS. Metabolic syndrome and an elevated plasma level of lipoprotein(a) have also
been associated with increased risk of calcific AS. The pathobiology of calcific AS is complex and
involves genetic factors, lipoprotein deposition and oxidation, chronic inflammation, osteoblastic
transition of cardiac valve interstitial cells and active leaflet calcification. Although no
pharmacotherapy has proved to be effective in reducing the progression of AS, promising therapeutic
targets include lipoprotein(a), the renin–angiotensin system, receptor activator of NF-
κB ligand
(RANKL; also known as TNFSF11) and ectonucleotidases. Currently, aortic valve replacement (AVR)
remains the only effective treatment for severe AS. The diagnosis and staging of AS are based on the
assessment of stenosis severity and left ventricular systolic function by Doppler echocardiography,
and the presence of symptoms. The introduction of transcatheter AVR in the past decade has been a
transformative therapeutic innovation for patients at high or prohibitive risk for surgical valve
replacement, and this new technology might extend to lower-risk patients in the near future.
significant cardiac blood flow obstruction (aortic jet velocity of <2.0 m per s)3. The prevalence of aortic
sclero-sis increases sharply with age. In developed countries, it is estimated to be 25% in those >65 years old and almost 50% in those aged >85 years9–11. According to a recent
meta-analysis, the rate of progression to AS in individ-uals with aortic sclerosis is 1.8–1.9% of patients per year11.
Therefore, the prevalence of calcific AS is much lower than that of aortic sclerosis, and has been estimated to be 0.4% in the general population and 1.7% in the popula-tion >65 years of age12 in developed countries. There is a
marked increase in the prevalence of calcific AS in those >65 years, as reported by several population-based studies in the United States and Europe9,13–15(FIG. 1).
For individ-uals aged ≥75 years, a pooled analysis of available epi-demiological data in developed countries produced an estimated severe AS prevalence of 3.4% (95% confidence interval of 1.1–5.7%), with 75% of those with severe AS presenting with symptoms16. The incidence of calcific
AS has been assessed in a longitudinal Norwegian study and was estimated to be 4.9 per 1,000 people per year in a population that had a mean age of 60 years at inclusion13.
The geographical distribution of calcific AS is hetero-geneous and shows a clustering effect, which is probably the consequence of genetic factors17.
Although mitral valve regurgitation has a higher prev-alence than AS in population-based studies, AS has a more important clinical effect18. In the Euro Heart Survey,
AS was more prevalent than mitral valve regurgitation in patients who were referred for in-hospital care and car-diac surgery18. Furthermore, calcific AS accounted for
34% of all native (that is, non-prosthetic) valve diseases, whereas mitral regurgitation accounted for 25%; and calcific AS accounted for 47% of patients operated for valvular disease, whereas mitral regurgitation accounted for 14% (REF. 18).
The burden of calcific AS in the community is expected to increase over the next decades owing to population ageing and the lack of a prevention strategy to reduce disease progression. Estimates based on current prevalence rates and demographic forecasts predict that the number of patients with calcific AS who are >70 or >75 years of age will increase twofold to threefold over the next 50 years in developed countries15,16,19.
The epidemiology of AS in developing countries and resource-poor settings differs in some respects to that seen in developed countries, partly because of the higher rates of rheumatic fever and rheumatic heart disease in poorer communities. Rheumatic heart disease is a chronic condition resulting from acute rheumatic fever, which in turn is caused by an untreated throat infection with group A Streptococcus. Both rheumatic fever and rheumatic heart disease may cause damage to the heart valves and can result in stenosis and regurgitation, particularly of the mitral and aortic valves. Valvular remodelling markedly differs between rheumatic heart disease and calcific AS. Fusion of aortic leaflets at commissures is one hallmark and distinctive feature of rheumatic heart disease; a dis-ease that rarely affects the aortic valve alone (less than 10% of all cases of valvular heart disease in countries in which rheumatic fever remains endemic) and most often involves the mitral valve. When the aortic valve is affected, the dysfunction is often mixed: aortic stenosis combined with some degree of aortic regurgitation20,21. The
propor-tion of AS caused by calcific AS is expected to increase in industrially developing countries owing to the decreas-ing incidence of rheumatic fever. In addition, the overall burden of calcific AS is expected to increase owing to the increase in life expectancy in these regions.
Mechanisms/pathophysiology
For a long time, calcific aortic valve disease was thought to be a ‘degenerative’ process caused by time-dependent wear and tear of the leaflets and passive calcium deposition. There are now compelling histopathological and clinical data suggesting that calcific valve disease is, in fact, an active and multifaceted condition involving lipoprotein deposition, chronic inflammation, osteoblastic transition of valve interstitial cells and active leaflet calcification22,23. Aortic valve anatomy and remodelling
The aortic valve is typically composed of three leaflets that are named according to their location with respect to the coronary artery; specifically, the left coronary, right coro-nary and non-corocoro-nary leaflets (FIG. 2). Each leaflet has a trilaminar structure that determines the biomechanical properties of the aortic valve24. The outermost layers of
the leaflet are formed by the fibrosa and ventricularis, which face the aorta and the left ventricular outflow tract, respectively. The spongiosa, which has a high proteoglycan content, is located between the fibrosa and ventricularis (FIG. 3). The fibrosa is rich in circumferentially oriented collagen type I and III fibres25, whereas in the ventricularis,
radially oriented elastic fibres predominate. The ventricu-laris composition provides compliance (that is, the ability to expand under pressure) and allows the apposition of free edge regions of leaflets, thus preventing the back-wards flow of blood into the left ventricle during diastole. The cellular population of these aortic valve layers includes valve interstitial cells (VICs), smooth muscle cells (SMCs; <5% of the population) and endothelial cells. The endothe-lial cells cover the aortic and ventricular surface and there-fore provide an interface between the blood and the aortic valve26. VICs are the predominant cells in the aortic valve,
whereas SMCs reside at the base of the ventricularis27.
Author addresses
1Cardiovascular Division, Washington University School of Medicine, St. Louis, Missouri, USA.
2Québec Heart and Lung Institute, Department of Medicine, Laval University, 2725 Chemin Sainte-Foy, Québec City, Québec G1V 4G5, Canada.
3Cardiology Department, AP-HP, Bichat Hospital, Paris, France.
4Paris-Diderot University, DHU Fire, Paris, France. 5University of Liège Hospital, GIGA Cardiovascular Sciences, Department of Cardiology, Heart Valve Clinic and CHU Sart Tilman, Liège, Belgium.
6Grupo Villa Maria Care and Research, Anthea Hospital, Bari, Italy.
7Division of Cardiology, Department of Medicine, University of Washington School of Medicine, Seattle, USA.
Inspection of surgically explanted valves with calcific AS reveals two features, fibrosis and calcification (FIG. 3), which substantially alter the biomechanical properties of the aortic valve leaflets. A small proportion (10–15%) of calcific AS valves show advanced osteogenic metaplasia with the presence of osteoblast-like cells, chondrocytes and bone marrow28. Calcified valves often contain dense
inflammatory infiltrates, which mostly consist of mac-rophages29,30. Mineralization starts in the fibrosa layer
and is often localized in the vicinity of lipid deposits. Together, these observations suggest that the fibro- calcific process in the aortic valve is a response to injury, which might be triggered by lipid-derived species and inflammation31(FIG. 4).
In addition, excess production and disorganization of collagen fibres is an important feature of calcific AS. Fibrosis increases the stiffness of the aortic valve and might play a considerable part in promoting mineral-ization. To this effect, the collagen produced by VICs functions as a scaffold on which the nucleation of calcium and phosphorus can start32. Serum-induced
mineralization of collagen is increased in vitro by a popu lation of VICs that have a pro-calcifying phenotype with elevated alkaline phosphatase (ALP) expression33,34.
Furthermore, the increased production of several com-ponents of the extracellular matrix, including periostin, tenascin (also known as tenascin C) and proteoglycans contributes to the remodelling of the aortic valve dur-ing AS35,36. The exact role of non-collagenous proteins
in the pathophysiology of AS is still mostly unknown, but growing evidence indicates that complex inter-actions between extracellular matrix proteins and cells provide crucial signals during normal reparative and pathological processes in the aortic valve37.
Lipids
Lipid infiltration and oxidation. Increasing evidence suggests that infiltration of the aortic valve by lipo-proteins has a central role in promoting inflammation, which precedes the pathological mineralization that is characteristic of calcific AS38. Therefore, the retention
of lipids promotes a chronic low-grade inflammatory Table 1 | Disease progression stages in calcific AS
Disease
stage Substage Description Management*
At risk of AS NA • Bicuspid aortic valve (or other congenital valve anomaly) or aortic valve sclerosis
• No obstruction to blood flow
• No symptoms
• Clinical and echocardiographic follow‑up every 3–5 years
• No indication of AVR Mild or
moderate AS NA • Mild-to-moderate leaflet calcification of a bicuspid valve or tricuspid valve with some reduction in systolic motion
• Mild or moderate AS‡
• Early left ventricular diastolic dysfunction might be present but normal LVEF
• No symptoms
• Clinical and echocardiographic follow‑up every 3–5 years for mild AS and every 1–2 years for moderate AS • No indication of AVR Severe AS Asymptomatic severe AS with normal left ventricular systolic function
• Severe leaflet calcification or congenital stenosis with a severely reduced leaflet opening
• Severe AS‡
• Left ventricular diastolic dysfunction but normal LVEF
• No symptoms
• Clinical and echocardiographic follow‑up every 6–12 months
• Indication of AVR (class IIa) if stenosis is very severe‡ and low surgical risk Asymptomatic severe AS with left ventricular systolic dysfunction
• Severe leaflet calcification or congenital stenosis with a severely reduced leaflet opening
• Severe AS‡ • LVEF of <50%
• No symptoms
• Indication of AVR (class I)
Symptomatic severe high-gradient AS
• Severe leaflet calcification or congenital stenosis with a severely reduced leaflet opening
• Severe AS with high gradient‡
• Left ventricular diastolic dysfunction, impaired left ventricular longitudinal systolic function and pulmonary hypertension may be present
• Symptoms include exertional dyspnoea, angina, syncope or pre-syncope and decreased exercise tolerance
• Indication of AVR (class I)
Symptomatic low-flow, low-gradient severe AS with preserved LVEF
• Severe leaflet calcification or congenital stenosis with a severely reduced leaflet opening
• Severe AS with low gradient‡
• Small left ventricular cavity with pronounced concentric remodelling, restrictive diastolic filling, and low-flow but normal LVEF
• Symptoms include heart failure, angina, syncope or pre-syncope
• Indication of AVR (class IIa)
Symptomatic low-flow, low-gradient severe AS with reduced LVEF
• Severe leaflet calcification or congenital stenosis with a severely reduced leaflet opening
• Severe AS with low gradient‡
• Left ventricular diastolic dysfunction and LVEF of <50%
• Symptoms include heart failure, angina, syncope or pre-syncope
• Indication of AVR (class IIa or class IIb if no left ventricular flow reserve)
AS, aortic stenosis; AVR, aortic valve replacement; LVEF, left ventricular ejection fraction; NA, not applicable. *Indication of AVR: for class I AVR should be carried
process that, in turn, might induce an osteogenic pro-gram in aortic valves. In this regard, histological studies have shown that several apolipoproteins (apos), such as apoB, apoE, apoA1 and apo(a), are present in surgically removed stenotic aortic valves39.
Oxidative stress has also been implicated in calcific AS. For instance, immunostaining has shown that apoB colo-calizes with oxidized low-density lipoproteins (Ox-LDLs) in valves from patients with calcific AS40,41, and that there
is an association between the level of Ox-LDL and the degree of inflammation and fibro-calcific remodelling in surgically removed AS valves40,42. Oxidative stress is
increased in AS valves and is at least partly related to the uncoupling of the nitric oxide synthase (NOS) path-way43. In addition, the expression NAD(P)H oxidase is
increased in surgically explanted calcific AS valves and contributes to the production of reactive oxygen spe-cies (ROS)44. Therefore, the production of peroxide and
superoxide anions in the vicinity of calcified areas might participate in the production of oxidatively modified lipid species with osteogenic properties43. Work carried
out in vitro has shown that Ox-LDL and several oxidized phospholipid (Ox-PL) species promote the calcification of isolated vascular cells45. Circulating Ox-PLs are mostly
carried in vivo by lipoprotein(a) (Lp(a))46, which is an
LDL-like particle in which the apoB protein is linked by a disulphide bridge to apo(a)47. Recent studies that used
a Mendelian randomization design showed that the gene encoding apo(a) (LPA) is potentially causally related to calcific aortic valve disease48–50. In addition, Capoulade
and colleagues showed that circulating Lp(a) and Ox-PL levels were independently associated with faster progres-sion of calcific AS51. Together, these studies suggest that
high circulating levels of Lp(a) might promote the accu-mulation of Ox-PLs in the aortic valve, which could, in turn, trigger an osteogenic response (FIG. 4).
Lipid retention and enzymatically modified lipid species. Proteoglycans such as biglycan and decorin are overexpressed in aortic valves during calcific AS and might actively participate in lipid retention and mod-ification52–54(FIG. 4). Moreover, transforming growth
factor β1 (TGFβ1), which is activated in calcific AS, has been shown to promote the elongation of glycosamino-glycan (GAG) chains55. In turn, GAG chain elongation
increases the interaction between proteoglycans and lipoproteins55. The accumulation and retention of
lipo-proteins in the aortic valve is a crucial event as lipids might be used by different enzymes to produce bioactive lipid-derived compounds, such as lysophospholipids56.
Lipoprotein-associated phospholipase A2 (Lp-PLA2)
levels are increased in stenotic aortic valves and this increase is associated with fibro-calcific remodelling57,58
(FIG. 4). Circulating levels of Lp-PLA2 are also positively and independently related to the progression of calcific AS59. Lp-PLA
2 is transported by apoB-containing
lipo-proteins and is enriched in small, dense LDL and Lp(a)60.
Lp-PLA2 transforms Ox-PLs into
lysophosphatidylcho-line (lysoPC), which promotes the loss of mitochondrial membrane potential and apoptosis of VICs57,61. In
addi-tion, Bouchareb and colleagues62 recently showed that
ectonucleotide pyrophosphatase/phosphodiesterase fam-ily member 2 (ENPP2; also known as autotaxin), a lyso-phospholipase D, is probably transported into the aortic valve by Lp(a) and is also secreted by VICs in response to diverse stimuli, including tumour necrosis factor (TNF; also known as TNFα)62. Autotaxin transforms lysoPC
into lysophosphatidic acid (lysoPA). Of interest, in vitro knockdown of autotaxin prevents the mineralization of VICs induced by lysoPC, which suggests that lysoPA is probably the mediator that promotes osteogenic pro-gramming in VICs. To this effect, in a mouse model, the administration of lysoPA increased the deposition of hydroxyapatite (a form of calcium apatite) in the aor-tic valve and accelerated the development of calcific AS. Therefore, it is possible that autotaxin and lysoPA are key factors that explain the link between Lp(a) and AS63.
In addition to lysophospholipids, the arachidonic acid pathway, which produces leukotrienes and prosta-glandins, has been shown to play a considerable part in the mineralization of the aortic valve64(FIG. 4). For
instance, the expression of 5-lipoxygenase, which is required for leukotriene synthesis, is increased in aortic valves during calcific AS, and leukotriene C4promotes the expression of bone morphogenetic protein 2 (BMP2) and BMP6 as well as the mineralization of VICs in cul-ture64. A recent study showed that prostaglandin G/H
synthase 2 (PTGS2; also known as cyclooxygenase 2 (COX2)) is expressed by VICs isolated from AS valves65.
In support of a role for COX2 in calcific AS, loss of func-tion of Cox2 in klotho-deficient mice, which develop
Nature Reviews | Disease Primers
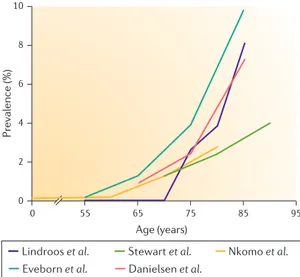
Pr evalence (%) 10 8 6 4 2 0 65 95 55 75 Age (years) 0
Lindroos et al. Stewart et al. Nkomo et al.
85
Eveborn et al. Danielsen et al.
Figure 1 | The prevalence of AS as a function of age.
The prevalence of aortic stenosis (AS) according to age in the following population-based series from the USA or Europe: Lindroos et al. (Finland)14, in which AS was defined as an aortic valve area of <1.2 cm²; Stewart et al.
(Cardiovascular Health Study, USA)9, in which AS was defined as a peak aortic jet velocity of >2.5 m per s; Nkomo et al. (USA)12, in which AS was defined as an aortic valve area of <1.5 cm2; Eveborn et al. (Tromsø Study, Norway)13, in which AS was defined as a mean gradient of ≥15 mm Hg; and Danielsen et al. (AGES-Reykjavik Study, Iceland)15, in which AS was defined as an indexed aortic valve area of ≤0.6 cm² per m².
calcification of the aortic valve amongst other features, reduced the mineralization of the aortic valve65. Taken
together, these findings suggest that several processes promote the retention of lipids in the aortic valve and produce bioactive lipid species, which in turn promote inflammation and mineralization of aortic valve leaflets.
Inflammation
Tissue remodelling and neovascularization. Fibro-calcific remodelling and inflammation of the aortic valve are intricately linked processes with important crosstalk. Inflammatory infiltrate in mineralized aortic valves that have been removed surgically is composed of macrophages, mast cells, CD4+ T cells and CD8+ T cells66.
Several oxidized lipid species might activate the innate immune response through Toll-like receptors (TLRs) and the nuclear factor-κB (NF-κB) pathway. TLRs are also expressed by VICs (in the case of TLR2 and TLR4) and may promote an osteogenic phenotype in isolated VICs67,68. Conversely, the role of adaptive immunity in
calcific AS is still mostly unknown, but studies have shown that a subset of memory T cells is activated during AS and that clonal expansion of a T cell receptor reper-toire is present in surgically removed calcific AS valves69.
These data suggest that both innate and adaptive immune responses are probably involved in the pathobiology of calcific AS.
A histopathological study carried out on 285 aortic valves from patients with calcific AS showed that the pres-ence of dense, chronic inflammatory infiltrates was related
to the remodelling score of the leaflets and to the presence of neovascularization29. Although the exact role of
neo-vascularization in driving AS is still mostly unknown, it is possible that it is involved in the recruitment of inflam-matory and osteoprogenitor cells (FIG. 4). In support of this hypothesis, mice that are deficient in chondromodulin 1 (encoded by Lect1), which is an anti-angiogenic factor, have thickened and mineralized aortic valve leaflets70.
Aged (22 months) Lect1−/− mice develop capillary-like
structures in their aortic valve leaflets, and this is accom-panied by the presence of inflammatory cells and lipid deposits70. In human stenotic aortic valves, CD34+
endothelial progenitor cells, which participate in new vessel formation, have been observed in clusters in close proximity to SPARC (also called osteonectin) and matrix metalloproteinase 9 (MMP9)71. SPARC is a matricellular
protein expressed by VICs during calcification that is cleaved by MMPs into peptides with angiogenic activity71.
Several MMPs, including MMP2, MMP9 and MMP12, are overexpressed in human calcific AS valve tissue72.
As such, angiogenic SPARC peptides might promote neo-vascularization by CD34+ endothelial progenitor cells and
might cause inflammation as well as remodelling of the aortic valve. In addition, cathepsins K, V and S, which are proteases that can degrade extracellular matrix proteins, are expressed and activated during AS73, and in ApoE−/−
mice, cathepsin S has been shown to promote elastolysis and mineralization of the aortic valve74. Therefore,
inflam-mation and neovascularization are linked to remodelling and mineralization of the aortic valve.
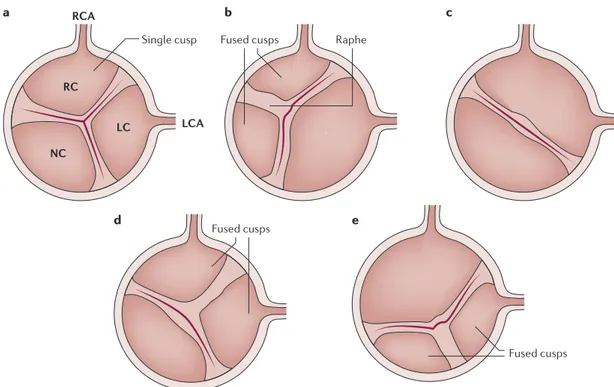
Nature Reviews | Disease Primers Single cusp RCA LCA RC LC NC
Fused cusps Raphe
a c
d e
b
Fused cusps
Fused cusps
Figure 2 | Comparison of tricuspid and bicuspid aortic valve structures. Schematic representation of a normal —
tricuspid — aortic valve with the three cusps (part a), a bicuspid valve with right non-coronary cusp fusion and one raphe
(the line of union between the fused cups) (part b), a bicuspid valve with fusion of the right and left coronary cusps and no
raphe (part c), a bicuspid valve with right–left coronary cusp fusion and one raphe (part d), and a bicuspid valve with fusion
of the left and non-coronary cups and one raphe (part e). LC, left coronary; LCA, left coronary artery; NC, non-coronary;
Cytokines. TNF is secreted by monocytes and macro-phages, and activates TNF receptor superfamily member 1A (TNFR1). TNFR1 activation results in activation of NF-κB and its downstream targets, including IL-1β and IL-6) (REFS 75–78) (FIG. 4). These cytokines promote the mineralization of VICs and activate an osteogenic pro-gramme, which may involve the expression of homeobox protein MSX2 (REFS 75–78). To this effect, treatment of adventitial fibroblasts with TNF increased the expression of MSX2 through the production of ROS79. Mice that are
deficient in IL-1 receptor antagonist protein (IL-1RN; encoded by Il1rn) have higher plasma levels of TNF than wild-type mice and develop a thickening of the aortic valve78. However, Il1rn−/−Tnf−/− mice are protected and do
not develop a thickening of the aortic valve, which sug-gests that TNF plays an important part in promoting the remodelling of the aortic valve. In humans, the expres-sion of TNF ligand superfamily member 10 (TNF10; also known as TRAIL), which is a member of the TNF-related cytokines, is increased in calcific AS valves and promotes the mineralization of VIC cultures through death receptor 4 (REF. 80).
IL-6, another cytokine with pleiotropic activities, has been implicated in calcific AS. IL-6 is increased in human calcified stenotic valves and is secreted in large amounts by cultured human VICs when they are treated with an osteogenic medium81. In addition, knockdown of IL6
sub-stantially reduces the expression of BMP2 and the mineral-ization of VIC cultures81. Moreover, although it has not yet
been investigated in VICs, IL-6 induces the expression of receptor activator of NF-κB ligand (RANKL; also known as TNFSF11) in bone cells, which activates its cognate recep-tor RANK (also known as TNFRSF11A)82. Overexpression
of RANKL during calcific AS might have an important role in pathogenesis, as secreted RANKL activates VICs to pro-duce extracellular matrix83(FIG. 4). In support of this role,
the administration of osteoprotegerin (OPG; also known as TNFRSF11b), which is a decoy receptor for RANKL, to low-density lipoprotein receptor knockout (Ldlr−/−) mice
decreased calcification and the expression of osteogenic genes in aortic valves84. Of interest, in bone, RANKL is
expressed by osteoblasts and promotes the resorption of mineral by osteoclasts. Therefore, it is possible that a dysregulation of RANKL–RANK–OPG explains the link between osteoporosis and vascular and valvular calcifi-cation66. In this regard, several epidemiological studies
have underlined an association between osteoporosis and vascular and/or valvular calcification66,85–87.
Angiotensin II
Angiotensin-converting enzyme (ACE) and chymase are overexpressed in calcific AS valves and are involved in the production of angiotensin II88,89(FIG. 4). Chymase is
secreted by mast cells present in calcific AS valve tissues and converts angiotensin I into angiotensin II88. In
addi-tion, patients with calcific AS have elevated blood plasma levels of angiotensin II, which correlates with the valvular expression of TNF and IL-6 (REF. 90). Angiotensin II is a potent activator of the NF-κB pathway and promotes a strong fibrotic response in isolated cells. In mice, the administration of angiotensin II promotes fibrosis of the aortic valve91. Moreover, in a rabbit model of
hypercholes-terolaemia, the administration of olmesartan, which is an angiotensin receptor blocker (ARB), prevents the thick-ening of the aortic valve that normally develops in these rabbits92. Retrospective non-randomized studies have
reported that administration of ARBs, but not ACE inhib-itors, is associated with less fibro-calcific remodelling of aortic valve leaflets and slower progression of valve steno-sis93,94. Therefore, it is possible that a substantial amount of
angiotensin II is produced by chymase in the aortic valve, the effect of which is blocked downstream by ARBs but not by ACE inhibitors.
Mineralization
Osteogenic differentiation. The endothelium that covers the healthy aortic valve expresses several anti- osteogenic genes in a spatially distributed manner95. The
endothe-lium that covers the aortic side of leaflets shows less expression of anti-osteogenic genes compared with the endothelium on the ventricular side. For instance, aortic- side endothelium expresses lower levels of chordin and OPG, which are negative regulators of BMP2, BMP4 and RANKL. A potential explanation for this difference in expression could be shear stress. Oscillatory shear stress has been shown to modulate the expression of ~1,000 genes and ~30 microRNAs (miRNAs) in human primary cultures of aortic valve endothelial cells96. For
instance, the expression of miRNA-187, which promotes cell growth and proliferation, was increased when these cultures were exposed to oscillatory shear. Endothelial cells covering the fibrosa (facing the aorta) are exposed to low oscillatory shear stress compared with cells facing the left ventricle. Although the functional relevance of these findings remains to be fully investigated, shear stress might at least partly explain why the fibro-calcific process predominantly occurs in the fibrosa layer.
Nature Reviews | Disease Primers
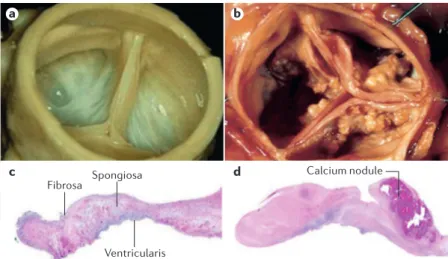
a c d b Fibrosa Spongiosa Ventricularis Calcium nodule
Figure 3 | Macroscopic and histopathological appearance of normal and abnormal aortic valves. Photographs of a normal aortic valve (part a) and an aortic valve with
severe calcific aortic stenosis (AS) (part b). Histopathological section of a normal aortic
valve with haematoxylin staining showing the trilaminar structure of the valve from top to bottom (part c). Histopathological section of a valve with severe calcific AS with
haematoxylin staining showing the presence of fibrotic material and a calcified nodule. The tissue is thickened by the excess of fibrotic material, and the calcified nodule, located in the fibrosa, contributes to alter the normal architecture of the leaflet (part d).
In human stenotic aortic valves, several osteogenic genes are overexpressed72, whereas others show altered
function that can affect their role in signalling path-ways. For instance, Garg and colleagues97 showed that
mutations in NOTCH1 were associated with bicuspid aortic valves, which are prone to developing calcific AS97. Notch family of receptors are involved in cell fate
determination. The activation of NOTCH1 in VICs leads to the formation of the Notch intracellular domain (NICD) fragment, which associates with the recombin-ing bindrecombin-ing protein suppressor of hairless (RBPJ) in the nucleus, where it promotes the expression of the hairy repressors. The hairy repressors prevent the expres-sion of the osteogenic factors BMP2 and runt-related
Leukotrienes
Nature Reviews | Disease Primers
Lipid infiltration Inflammation Fibro-calcific response
Radiation Mechanical stress Lipid-derived species Cytokines VIC Ox-LDL Ox-PL Angiotensin I Angiotensin II RANKL TNF LPAR ATX ENPP1 Fibrosis NT5E BMP2 AA Prostaglandins sPLA2 MMPs ATX Lp(a) LDL NOS uncoupling ROS lysoPC Lp-PLA2 lysoPA LDL ACE Chymase Mastocyte T cell Monocyte Macrophage Calcifying microvesicles VEGF TNFIL-1β Collagen Mineralization RUNX2 MSX2 Inflammation VEGF Lipids Calcium hydroxyapatite Osteoprogenitor cell Apoptosis Osteogenic transition TGFβ WNT3a IL-6 5-LO COX2 A2AR ATP AMP +PPi Pi ALP Adenosine +Pi Blood vessel Time
Figure 4 | Pathogenesis of calcific AS. Endothelial damage allows infiltration
of lipids, specifically low-density lipoprotein (LDL) and lipoprotein(a) (Lp(a)) into the fibrosa and triggers the recruitment of inflammatory cells into the aortic valve. Endothelial injury can be triggered by several factors including lipid-derived species, cytokines, mechanical stress and radiation injury. The production of reactive oxygen species (ROS) is promoted by the uncoupling of nitric oxide synthase (NOS), which increases the oxidation of lipids and further intensifies the secretion of cytokines. Enzymes transported in the aortic valve by lipoproteins (that is, LDL and Lp(a)) such as lipoprotein-associated phospholipase A2 (Lp-PLA2) and ectonucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2; also known as autotaxin (ATX)) produce lysophospholipid derivatives. ATX, which is also secreted by valve interstitial cells (VICs), transforms lysophosphatidylcholine (lysoPC) into lysophosphatidic acid (lysoPA). Several factors including lysoPA, the receptor activator of nuclear factor-κB ligand (RANKL; also known as TNFSF11) and WNT3a promote the osteogenic transition of VICs. Arachidonic acid (AA) generated by cytosolic PLA2 promotes the production of eicosanoids (for example, prostaglandins and leukotrienes) through prostaglandin G/H synthase 2 (PTGS2; also known as COX2) and 5‑lipoxygenase (5‑LO) pathways, respectively. In turn, eicosanoids promote inflammation and mineralization. Chymase and angiotensin-converting
enzyme (ACE) promote the production of angiotensin II, which increases the synthesis and secretion of collagen by VICs. Owing to increased production of matrix metalloproteinases (MMPs) and decreased synthesis of tissue inhibitors of metalloproteinases (TIMPs), disorganized fibrous tissue accumulates within the aortic valve. Microcalcification begins early in the disease, driven by microvesicles secreted by VICs and macrophages. In addition, overexpression of ectonucleotidases (ENPP1, 5ʹ-nucleotidase ecto (NT5E)) and alkaline phosphatase (ALP)) promotes both apoptosis and osteogenic-mediated mineralization. Bone morphogenetic protein 2 (BMP2) leads to osteogenic transdifferentiation, which is associated with the expression of bone-related transcription factors (for example, runt-related transcription factor 2 (RUNX2) and homeobox protein MSX2). Osteoblast‑like cells subsequently coordinate calcification of the aortic valve as part of a highly regulated process analogous to skeletal bone formation. Deposition of mineralized matrix is accompanied by fibrosis and neovascularization, which is abetted by vascular endothelial growth factor (VEGF). In turn, neovascularization increases the recruitment of inflammatory cells and bone marrow-derived osteoprogenitor cells. A2AR, adenosine A2A receptor; sPLA2, secreted PLA2; LPAR, lysophosphatidic acid receptor; Ox‑PL, oxidized phospholipid; Ox‑LDL, oxidized LDL; TGFβ, transforming growth factor-β; TNF, tumour necrosis factor.
transcription factor 2 (RUNX2) in VICs98, suggesting
that VICs are driven towards an osteogenic differentia-tion pathway in calcific AS. To this effect, heterozygous Notch1+/− and Rbpj+/− mice develop mineralization of
the aortic valve99. In addition, the NICD interferes in
the nucleus with β-catenin (also known as catenin β1), a downstream effector of the WNT pathway, which is also a key driver of osteogenic differentiation100. A recent
study in endothelial cells showed that NOTCH1 regu-lates the expression of more than a 1,000 genes involved in inflammation and osteogenesis by altering the epi-genetic signature at enhancer regions101. Moreover, in
human stenotic aortic valves, WNT3a, an agonist of the WNT pathway, is overexpressed102. The activation
of a co-receptor formed by LDLR-related protein 5 and G protein-coupled Frizzled receptors, which are expressed by VICs, leads to the stabilization of β-catenin and to osteogenic differentiation102(FIG. 4). In vascular
cells, BMP2 promotes the expression of MSX2, a pos-itive regulator of the WNT pathway103. Several factors,
including inflammatory cytokines and oxidized lipid derivatives, have been shown to induce the expression of BMP2 in different cell types, including VICs104.
Recent studies have also highlighted that the expres-sion of several miRNAs is dysregulated in AS and this might affect the osteogenic programming of VICs. In this regard, miRNA-30b, which is decreased in min-eralized aortic valves, is a negative regulator of RUNX2 (REF. 105). Hence, a dysfunction of Notch and WNT path-ways as well as a dysregulation of miRNAs contribute to increased pro-osteogenic signals in VICs.
Mineral deposition. Osteogenic reprograming of VICs brings about a series of events that promote the depo-sition of a calcified matrix. The mechanism (or mech-anisms) by which VICs mineralizes the extracellular matrix is still poorly defined but recent observational and experimental work suggests that cells secrete small vesicles rich in ectonucleotidases that promote the nucleation of calcium and phosphorus106,107. A build-up
of phosphate in calcifying vesicles, which also contain the annexin V–S100A9 complex that binds calcium, promotes the nucleation of minerals108. Secretion of
calcifying vesicles has classically been attributed to cells that transdifferentiate into osteoblast-like cells, in which case calcification proceeds with the deposition of well- organized bone-like mineral matrix (known as hydroxyapatite of calcium)109. However, programmed
cell death leads to the production of apoptotic bodies with similar properties to calcifying vesicles. Apoptosis of VICs is promoted by different stimuli including cytokines, ROS and altered purinergic signalling. Apoptotic bodies function as nidi for dystrophic calci-fication, a form of mineralization that consists of amor-phous deposits of calcium and phosphorus crystals. In human aortic valves, it is likely that both osteogenic and apoptotic processes contribute to the mineralization process, and at least partly rely on ectonucleotidases110.
In support of this involvement, several ectonucleotidases, such as ALP, ENPP1 and 5ʹ- nucleotidase (5ʹ-NT; also known as CD73), are overexpressed in human stenotic
aortic valves110–112 (FIG. 4). These membrane-bound
enzymes use nucleotides and nucleosides secreted by cells as substrates and produce phosphate-derived prod-ucts that promote mineralization112. For instance, ENPP1
hydrolyses ATP into AMP and pyrophosphate, which is a strong inhibitor of mineralization. Conversely, ALP has a broad range of substrates, including the mineralization inhibitor pyrophosphate from which it produces phos-phate with strong pro-mineralizing activity. Moreover, the overactivity of ENPP1 and 5ʹ-NT in human stenotic aortic valves depletes extracellular ATP and produces adenosine with osteogenic activity111. A decrease in
the level of extracellular ATP also diminishes puriner-gic signalling through the P2Y purinoceptor 2 (P2Y2). In VICs, P2Y2 prevents the mineralization of cells by interfering with apoptosis and also by promoting the activation of carbonic anhydrase 12 (CA12)110,113. CA12
in VICs is normally expressed at the cell membrane fol-lowing activation of P2Y2 and promotes the acidification of the extracellular space leading to resorption of min-eral deposits113. As such, purinergic signalling, which is
under the control of ectonucleotidases, plays a central part in controlling the mineralization of the aortic valve.
In summary, studies carried out in the past several years have shown that oxidation and infiltration of the aortic valve by lipids generate several bioactive lipid spe-cies that trigger inflammation of the aortic valve. The activation of several pathways with multiple points of crosstalk disrupts the normal biology of the aortic valve and promotes fibro-calcific remodelling.
Pathophysiology of left ventricular dysfunction
The symptoms in AS are essentially due to an imbalance between the increase in left ventricular haemodynamic load caused by valvular obstruction, on the one hand, and the capacity of the left ventricle to overcome this increase in load both at rest and during exercise, on the other hand. AS results in increased left ventricular sys-tolic pressure that leads to hypertrophy of the cardiomy-ocytes and interstitial fibrosis (FIG. 5). The mechanical signal generated by increased left ventricular systolic pressure initiates a cascade of biological events, includ-ing re-expression of immature fetal genes, which leads to coordinated cardiac growth in patients with AS114. This
increase in cardiac mass is due to the hypertrophy of existing myocytes rather than to hyperplasia, because cardiomyocytes become terminally differentiated soon after birth. The concurrent addition of sarcomeres (force-generating units) causes an increase in myocyte width, which in turn increases wall thickness and there-fore contributes to normalization of left ventricular wall stress and maintenance of left ventricular ejection per-formance despite elevated systolic pressure. To support the increased biomechanical load, the myocyte growth must be accompanied by coordinated increases in the surrounding architecture of connective tissue as well as the capillary and nerve networks114. This ‘reactive’
inter-stitial fibrosis that results from the increase in collagen synthesis by myofibroblasts in response to pressure over-load has a diffuse distribution within the interstitium and might be, at least partly, reversible following AVR115.
The pattern of the left ventricular adaptive response to pressure overload in AS is highly heterogeneous and includes concentric remodelling, concentric hyper-trophy and eccentric hyperhyper-trophy (FIG. 6). The pattern and magnitude of left ventricular hypertrophic remod-elling is influenced not only by AS severity but also by several other factors, including age, sex, genetic factors, metabolic factors and the coexistence of coronary artery disease or hypertension116–119. Among individuals with
the same degree of AS, women tend to develop concen-tric remodelling or concenconcen-tric hypertrophy most often, whereas men are more prone than women to develop-ing eccentric hypertrophy116. In patients with calcific
AS, left ventricular concentric remodelling or hypertro-phy has been linked to worse myocardial function and increased risk of cardiac events and mortality compared with patients with normal left ventricular geometry or with left ventricular eccentric hypertrophy120–122. Obesity,
metabolic syndrome and diabetes also predispose an individual to the development of more concentric hypertrophy in the presence of AS117,118.
The left ventricular hypertrophy that leads to a reduced density of coronary arteriolar vessels, and the increased left ventricular transmural pressures that lead to increased coronary vascular resistance, result in the reduction of coronary flow reserve in patients with AS123,124. The reduction of coronary flow reserve
limits the ability of the coronary circulation to increase flow to match myocardial oxygen demand, especially during exercise, and it is therefore a key factor in the development of myocardial ischaemia and the occur-rence of symptoms. Repetitive myocardial ischaemia related to the exhaustion of coronary flow reserve leads
to apoptosis of myocytes and to the development of ‘replacement’ myocardial fibrosis. This type of fibrosis occurs predominantly in the subendocardial and mid-wall layers of the left ventricle mid-wall and is generally not reversible following relief of left ventricular pressure overload by AVR. The impairment of coronary flow reserve might also explain why patients with severe AS can present with angina symptoms despite having angio-graphically normal coronary arteries, and why these symptoms might regress immediately after AVR125.
Left ventricular diastolic dysfunction occurs early in the disease course and worsens with progression of stenosis severity and myocardial fibrosis (FIG. 5). In the more advanced stages of the disease, the increased left ventricular filling pressures lead to secondary pulmo-nary hypertension and dyspnoea symptoms126,127. The
global left ventricular systolic function, which is meas-ured using the left ventricular ejection fraction (LVEF), and cardiac output are generally well preserved even in the presence of severe AS, because the increase in left ventricular wall thickness allows wall stress to remain relatively normal. Reduced LVEF or cardiac output occurs only in end-stage disease and is usually preceded by clinical symptoms. However, a large proportion of patients with preserved LVEF have subtle left ventricular systolic dysfunction that is characterized by impaired left ventricular longitudinal function with relatively well pre-served radial and circumferential function (BOX 1). The left ventricular myocardial wall is composed of three lay-ers from the inside to the outside of the left ventricle: the subendocardial layer that surrounds the left ventricular cavity, the mid-wall layer and the subepicardial layer. In pressure overload cardiomyopathies, there is an early Figure 5 | Maladaptive remodelling and impaired function of the left ventricle in response to pressure overload from AS. The narrowing of the aortic valve orifice causes an acceleration of the blood flow velocity with a concomitant
decrease in systolic blood pressure between the left ventricular outflow tract (LVOT) and the aorta. The increased left ventricular pressure imposed by AS results in left ventricular hypertrophy (augmentation of the left ventricular myocardial mass), reduced coronary flow reserve, myocardial fibrosis, diastolic dysfunction and decreased longitudinal systolic shortening, although the ejection fraction remains normal in most patients. Left atrial enlargement is common owing to elevated left ventricular filling pressures, which often lead to secondary pulmonary hypertension and right ventricular dysfunction in the more advanced stages of the disease.
Nature Reviews | Disease Primers Calcified aortic valve
Left ventricular hypertrophy Reduction in coronary flow reserve Myocardial fibrosis Systolic blood pressur
e (mm
Hg)180
120
Aortic valve Aorta LVOT Blood velocity (m/s) 1 4 Anatomical location Pulmonary hypertension Diastolic dysfunction
Reduced left ventricular longitudinal systolic function
and selective alteration of the shortening of myocardial fibres within the subendocardial layer in which ischae-mia and fibrosis are generally more pronounced128–130
(FIG. 5). The fibres in this layer are oriented longitudinally (compared with circumferentially in the mid-wall layer), which explains the selective alteration of the left ven-tricular longitudinal function in these patients. Hence, a considerable proportion of patients with AS may have subclinical left ventricular systolic dysfunction despite preserved LVEF and the absence of symptoms.
Diagnosis, screening and prevention Risk factors and prevention
Although some clinical and genetic risk factors have been associated with the onset and progression of calcific AS, no strategy has so far been proved to be efficient for primary or secondary prevention of this disease. Calcific AS shares several risk factors with cor-onary artery disease but it also presents some important distinctive features.
Clinical risk factors. Congenital leaflet abnormality and older age are both powerful risk factors for devel-oping calcific AS. For instance, the lifetime risk of AVR is approximately 50% in individuals with a bicus-pid valve. Bicusbicus-pid aortic valves have two functional
leaflets often of unequal size. This abnormality results from incomplete separation of commissures during embryonic development8. Although leaflet orientation
varies among patients, the most common form con-sists of a fusion of the right and left coronary leaflets (in ~60% of patients), followed by fusion between the right and the non- coronary leaflets (in ~35% of patients), and fusion between left and non-coronary cusp (in ~5% of patients)131(FIG. 2). A bicuspid aortic valve is associated
with an increased risk of aortopathy, in which genetic, haemodynamic and mechanical factors might participate in the mineralization of the aortic valve132. In individuals
with a bicuspid valve and in those with a tricuspid valve, age is a powerful risk factor for AS9,133. The other clinical
risk factors associated with AS are similar to those associ-ated with atherosclerosis and include male sex, smoking, hypertension, hypercholesterolaemia, obesity, metabolic syndrome, diabetes and elevated Lp(a)9,48,134–136.
In patients with AS, the rate of stenosis progres-sion over time varies substantially from one patient to another. The clinical factors associated with faster steno-sis progression include older age, severity of the stenosteno-sis and the degree of aortic valve calcification at diagnosis, smoking, hypertension, obesity, metabolic syndrome, secondary hyperparathyroidism, renal failure, ele-vated circulating levels of Lp(a) and increased activity of Lp-PLA2(REFS 51,59,94,137–142). In particular, the
presence of elevated plasma Lp(a) (>50 mg per dl; the upper normal limit is 30 mg per dl) is associated with a twofold faster stenosis progression51.
In addition, hypertension, and particularly systolic hypertension, is highly prevalent in these patients, affect-ing 30–70% of those with AS94,143,144. Recent studies
sug-gest that hypertension accelerates the progression of AS, potentially owing to increased mechanical stress on the valve leaflets and activation of the renin–angiotensin system (as discussed above)94. Moreover, hypertension
further increases the left ventricular afterload (BOX 1) that is already elevated in patients with AS and contrib-utes to the risk of developing symptoms and adverse cardiac events94,144.
Genetic risk factors. Several studies suggest that a genetic component is involved in promoting calcific AS associ-ated with bicuspid or tricuspid aortic valves6,17,48,145.
However, despite the evidence of a strong inheritance pattern for some cases of bicuspid aortic valve with an incomplete penetrance, the genetic architecture of calcific AS is still poorly understood145. So far, variants
of NOTCH1 and GATA-binding protein 5 (GATA5) have been associated with bicuspid aortic valves in humans97,146,147. NOTCH1 mutations explain
approxi-mately 4% of sporadic cases of AS that occur in the con-text of a bicuspid aortic valve148,149. As discussed above,
some mutations in NOTCH1 that affect its function might promote aortic valve mineralization. Therefore, it is possible that gene variants that predispose individuals to developing a bicuspid aortic valve also promote valve mineralization later in life, thus further exacerbating the risk of developing calcific AS. A recent genome-wide association study found that variants located in RUNX2 Figure 6 | Patterns of left ventricular remodelling. Four left ventricular remodelling
patterns can be defined according to the left ventricular mass and the ratio of the left ventricular mass to the left ventricular cavity size: for normal pattern both left ventricular mass and mass/cavity ratio are normal; for concentric remodelling the left ventricular mass is normal but the mass/cavity ratio is increased (thick left ventricular walls with small cavity); for concentric hypertrophy both left ventricular mass and mass/cavity ratio are increased; and for eccentric remodelling left ventricular mass is increased but the mass/cavity ratio is normal (thickness of left ventricular walls is normal or slightly increased and the left ventricular cavity is enlarged). Figure is reproduced from REF. 267, Nature Publishing Group.
Nature Reviews | Disease Primers
Concentric remodelling Concentric hypertrophy
Normal Eccentric hypertrophy
Absent Incr eased Left v entricular mass/cavity r atio Normal Present Left ventricular hypertrophy
and calcium channel voltage-dependent L-type alpha 1C subunit (CACNA1C), which encode an osteogenic transcription factor and a voltage-dependent calcium channel subunit, respectively, were associated with cal-cific AS and were found to upregulate their respective mRNA levels150. Also, studies using a candidate gene
approach have linked several gene variants with calcific AS. Although variants of vitamin D receptor (VDR),
APOE, APOB, IL10, NOTCH1 and ENPP1 have been found to be significantly associated with AS, these stud-ies suffer from small sample size and require replication in larger series6.
A large study using a Mendelian randomization design identified the single-nucleotide polymorphism (SNP) rs10455872 in the LPA gene as the only genome-wide significant SNP associated with the presence of aortic valve calcification and clinical calcific AS48.
Subsequent studies have validated these findings and have also reported an association between elevated Lp(a) plasma levels and the prevalence of calcific AS, and the need for AVR in the general population49–51.
The presence of the rs10455872 allele is associated with a 1.5–2.0-fold increase in the risk of incident calcific AS48–50. When considered in light of the clinical and basic
research findings on Lp(a) discussed above, lowering of Lp(a) seems to be a promising novel target for the treat-ment of this disease, particularly to prevent disease pro-gression. However, further studies are needed to evaluate the role of Lp(a) in AS in more detail.
A second study using a Mendelian randomization design reported a strong association between genetic predisposition to elevated LDL cholesterol, as measured by weighted genetic risk scores, and the presence of aortic valve calcification and incident cases of calcific AS151.
However, three randomized clinical trials failed to show any significant benefit of lowering LDL using statins on the progression of AS152–154. Therefore, it is possible that
elevated LDL cholesterol promotes the initiation of cal-cific aortic valve disease but has minimal or no effect on AS progression. Moreover, the protective effect of statin therapy in AS might be counterbalanced by its off- target effects, including pro-osteogenic properties, worsening of insulin resistance and increased Lp(a) levels51,141.
Whether other lipid-lowering strategies (for instance, proprotein convertase subtilisin/kexin-type 9 (PCSK9) inhibitors) would prevent or slow AS progression is unknown and this question needs to be addressed. In summary, no pharmacotherapy has proved to be effective in reducing the progression of AS.
Diagnosis
Diagnosis of AS is generally established using an echo-cardiographic examination, which provides a wealth of information regarding heart valve anatomy and blood flow parameters155(FIG. 7). The same techniques
can be used for the diagnosis of calcific AS and rheu-matic AS. In the vast majority of patients, referral to echocardio graphy is motivated by the auscultation of a systolic murmur and/or the development of symptoms including dyspnoea, angina, syncope and dizziness. In some cases, AS is first recognized on echocardio-graphy requested for other indications. Although most patients are diagnosed long before the onset of symp-toms and are followed prospectively on a regular basis until AVR is indicated, a small proportion (5–10%) of patients are not diagnosed with AS until late in the disease course when they present with symptoms of heart failure156. The identification of the presence and
stage of AS includes the assessment of the aortic valve Box 1 | Key measurements and tools used for AS assessment
• Aortic valve area (AVA): surface of the aortic valve orifice. It can be measured by Doppler echocardiography, left heart catheterization or cardiac magnetic resonance.
• Aortic valve calcium density: aortic valve calcium score measured by CT divided by the cross-sectional area of the aortic annulus measured by echocardiography or CT. It is expressed in Agatston units per cm2.
• Carotid upstroke: the pulse pressure of the carotid artery that can be assessed at the level of the neck is characterized by a smooth, fairly rapid upstroke and a smooth, more gradual downstroke. In patients with severe aortic stenosis, the carotid upstroke is delayed.
• Circumferential function: circumferential contraction of the left ventricular wall that is mainly driven by the myocytes located in the mid portion of the left ventricular wall.
• Class of recommendation for the procedure (aortic valve replacement in the case of aortic stenosis (AS)): for class I the benefit of the procedure mainly outweighs the risk and the procedure should be carried out; for class IIa it is reasonable to carry out the procedure; for class IIb the procedure may be considered; and for class III the procedure is not recommended because it is not useful and may be harmful.
• Coronary flow reserve: the ratio of maximum blood flow through the coronary arteries compared with the normal resting flow. The coronary flow reserve can be measured by cardiac catheterization, Doppler echocardiography or positron emission tomography. The normal coronary flow reserve ratio is 3-4. In patients with AS, the coronary flow reserve is reduced. When the ratio is 1, the coronary flow reserve is exhausted.
• Dobutamine stress echocardiography: echocardiography carried out during intravenous infusion of dobutamine, which increases cardiac contractility and flow across the aortic valve.
• Mean transvalvular gradient (mean gradient): average value of the pressure loss (or gradient) across the aortic valve. This corresponds to the difference between the pressure in the left ventricular cavity versus that in the aorta. The mean gradient can be measured by Doppler echocardiography or by left heart catheterization.
• Left ventricular afterload: pressure in the wall of the left ventricle during ejection.
• Left ventricular ejection fraction (LVEF): measurement of how much blood is being pumped out of the left ventricle of the heart. It is calculated as the percentage decrease in the volume of the left ventricular cavity. It can be measured by echocardiography, angiography or cardiac magnetic resonance.
• Left ventricular longitudinal function: longitudinal (that is, long-axis direction) contraction of the left ventricular wall that is mainly driven by the myocytes located in the subendocardial layer of the left ventricular wall.
• Longitudinal strain: percentage shortening of the left ventricular wall in the longitudinal axis during systole. The longitudinal strain can be measured by speckle tracking echocardiography.
• Peak aortic jet velocity: peak value of the blood flow velocity across the aortic valve. The blood velocity is measured by continuous-wave Doppler.
• Radial function: longitudinal (that is, short-axis direction) contraction of the left ventricular wall that is mainly driven by the myocytes located in the mid-wall layer of the left ventricular wall.
• Stress AVA: AVA measured by Doppler echocardiography during dobutamine or exercise stress.
• Stress mean gradient: mean gradient measured by Doppler echocardiography during dobutamine or exercise stress.
• Stroke volume index: stroke volume (that is, volume of blood ejected by the heart during systole) indexed to (divided by) the patient’s body surface area.
anatomy and morphology, the haemodynamic severity of AS, the response of the left ventricle to the pressure overload caused by AS and the patient’s symptomatic status3,4. On the basis of these assessments, patients can
be diagnosed with mild, moderate or severe AS, which can all occur in the presence or absence of symptoms (TABLE 1). Although Doppler echocardiography is the primary modality to assess the stage of AS, cardiac cath-eterization, which can measure cardiac blood pressure and flow, may be used to confirm the haemodynamic severity of the stenosis in patients with inconclusive or discordant echocardiography results157. However, this
invasive technique is associated with increased risk of bleeding and cerebral embolism158, and should therefore
only be considered in patients in whom the reclassifi-cation of the stenosis severity by catheterization would change the therapeutic management of the patient (such as AVR versus conservative management). For exam-ple, individuals who might benefit from catheterization
assessment include symptomatic patients for whom a diagnosis of moderate AS versus severe AS cannot be decided using echocardiography.
Patients at risk for AS. Individuals with aortic sclero-sis and those with a bicuspid valve (irrespective of the presence or absence of sclerosis) are considered to be at risk of developing AS. The identification of a bicuspid valve is usually done by echocardiography but might require other imaging modalities such cardiac magnetic resonance (CMR) or CT if the valve is calcified.
Aortic valve sclerosis is defined echocardiographi-cally by focal areas of valve calcification and thicken-ing with normal leaflet mobility and normal valvular haemo dynamics (FIG. 7; TABLE 2). A systolic outflow mur-mur may be auscultated on physical examination. Although aortic sclerosis is clinically asymptomatic, its presence is independently associated with a 40% increase in the risk of a coronary event and a 50% increase in
Nature Reviews | Disease Primers
a c d b 0 –60 –120 –180 –240 0 –100 –200 –300 –400 0 –60 –120 –180 –240 –300 60 0 –100 –200 –300 –400 cm/s cm/s cm/s cm/s 100mm/s 100mm/s 100mm/s 53bpm 100mm/s 51bpm 53bpm 71bpm Aortic valve Aortic valve
Figure 7 | Assessment of AS severity by Doppler echocardiography. For each degree of disease severity, including
aortic valve sclerosis (part a), mild aortic stenosis (AS) (part b), moderate AS (part c) and severe AS (part d), this figure
shows a 2D echocardiographic short-axis view of the aortic valve (top left panel, indicated with arrows), the transvalvular velocity by continuous-wave Doppler (right panel) and the multidetector CT (MDCT) view of aortic valve calcification (bottom left panel). In the patient with aortic sclerosis (part a), there are some small isolated spots of calcification (appears
white on the MDCT images, circled) in the aortic valve leaflets but there is no obstruction to blood flow (that is, no stenosis). The peak aortic jet velocity (1.47 m per s), mean gradient (5 mm Hg) and aortic valve area (AVA; 2.87 cm2) are normal. In the patient with mild AS (part b), there is mild aortic valve calcification with mild obstruction to blood flow.
The peak aortic jet velocity is 2.08 m per s, the mean gradient is 9 mm Hg and the AVA is 1.62 cm2. In the patient with moderate AS (part c), there is more extensive aortic valve calcification with moderate obstruction of blood flow: the peak
aortic jet velocity is 3.51 m per s, the mean gradient is 28 mm Hg and the AVA is 1.21 cm2. In the patient with severe AS (part d), there is severe aortic valve calcification and severe obstruction to blood flow: the peak aortic jet velocity is 4.35 m
the risk of cardiovascular death159. The mechanism of
adverse outcomes with aortic sclerosis is not entirely clear but the presence of aortic valve mineralization might be a marker for atherosclerosis and/or for altered phospho-calcium metabolism22,160.
Mild or moderate AS. Patients with mild or moderate AS
(FIG. 7; TABLES 1,2) are generally asymptomatic unless they have other comorbidities that contribute to the emergence of symptoms. Classic physical findings of AS are a harsh, crescendo–decrescendo systolic murmur, a single sec-ond heart sound and a delayed carotid upstroke (BOX 1). Using Doppler echocardiography, the haemodynamic severity of AS can be measured accurately and reliably on the basis of the peak aortic jet velocity, mean trans-valvular pressure gradient (mean gradient) and aortic valve area (AVA). With the development of calcific AS, there is a progressive reduction in the AVA that causes an acceleration of the flow (that is, increase in peak aortic jet velocity) and a loss of pressure (that is, increase in mean gradient) across the valve (FIG. 6; TABLE 2). AS is suspected upon the visualization of a thickened aortic valve with a restricted opening, and confirmed by the presence of an increased peak aortic velocity or mean pressure gradient. Echocardiography is also useful to assess the effects of AS on the geometry and the function of cardiac chambers, particularly of the left ventricle (FIGS 5,6).
Severe AS. Patients with severe AS (typically, those who have a peak aortic jet velocity of ≥4 m per s, a mean gra-dient of ≥40 mm Hg and an AVA of ≤1 cm2; TABLES 1,2)
may or may not have symptoms, and require a closer clinical and Doppler echocardiographic follow-up than those with mild or moderate forms of the disaese3.
Classic symptoms of severe AS include dyspnoea and other symptoms of heart failure, angina and syncope. Patients with severe AS who are apparently asympto-matic according to medical history and physical exami-nation should undergo exercise testing to confirm their asymptomatic status. Indeed, about one-third of patients with severe AS who are a priori asymptomatic in fact have exercise-limiting symptoms detected at an exercise
stress test, and these patients should be referred for AVR161,162. In addition, a potential marker for risk in AS
is a marked increase in mean gradient (absolute increase in gradient >18–20 mm Hg) during exercise stress echo-cardiography, which predicts higher risk of cardiac events in the short term, independent of symptoms161,162.
Low-gradient AS. The majority of patients with severe AS have a high peak aortic jet velocity and gradient (mean gradient ≥40 mm Hg). However, a substantial propor-tion of patients may have a low peak aortic jet velocity and mean gradient despite the presence of a small AVA (<1.0 cm2). The most frequent cause of ‘low-gradient’
AS is the presence of a low-flow state. There are two main subtypes of low-flow, low-gradient AS (TABLES 1,2): ‘classical’ low-flow (stroke volume index <35 ml per m2), low-gradient (mean gradient <40 mm Hg) AS with
reduced LVEF (<50%)163; and ‘paradoxical’ low-flow
(stroke volume index <35 ml per m2), low-gradient (mean
gradient <40 mm Hg) AS with preserved LVEF (≥50%)164.
In classical low-flow, low-gradient AS, the decrease in stroke volume, and thus in transvalvular flow rate (stroke volume divided by left ventricular ejection time), are predominantly related to left ventricular systolic dys-function, whereas in paradoxical low-flow, low-gradient AS, the low-flow state is generally due to pronounced left ventricular concentric remodelling with impaired left ventricular diastolic filling and reduced left ventricular longitudinal systolic function156. Other conditions, such
as mitral regurgitation, mitral stenosis or atrial fibril-lation can also contribute to the reduced left ventricu-lar outflow in both classical and paradoxical low-flow, low-gradient AS.
In the presence of low flow, it is therefore difficult — using resting Doppler echocardiography or catheteri-zation — to differentiate truly severe stenosis from pseu-do-severe stenosis; that is, a situation in which the stroke volume is not sufficient to completely open a valve that is only mildly or moderately stenotic. In such low-flow conditions, the gradient might underestimate the steno-sis severity, whereas the AVA might overestimate the severity. Low-dose dobutamine stress echocardiography Table 2 | Parameters and criteria for the assessment of aortic stenosis severity
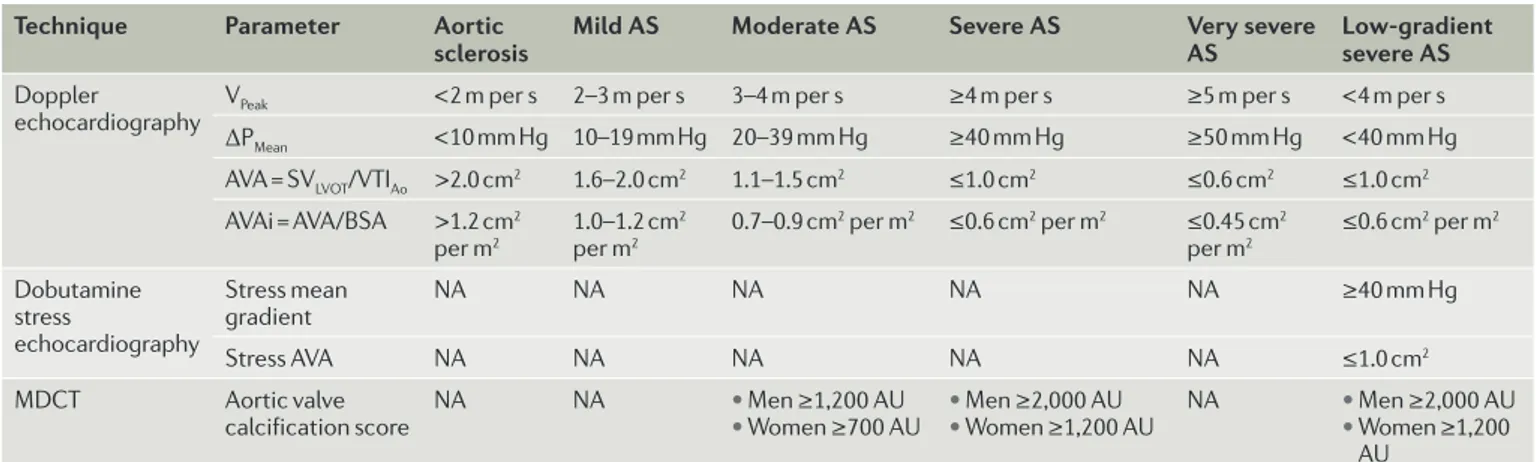
Technique Parameter Aortic
sclerosis Mild AS Moderate AS Severe AS Very severe AS Low-gradient severe AS
Doppler
echocardiography VPeak <2 m per s 2–3 m per s 3–4 m per s ≥4 m per s ≥5 m per s <4 m per s ΔPMean <10 mm Hg 10–19 mm Hg 20–39 mm Hg ≥40 mm Hg ≥50 mm Hg <40 mm Hg AVA = SVLVOT/VTIAo >2.0 cm2 1.6–2.0 cm2 1.1–1.5 cm2 ≤1.0 cm2 ≤0.6 cm2 ≤1.0 cm2 AVAi = AVA/BSA >1.2 cm2 per m2 1.0–1.2 cm 2 per m2 0.7–0.9 cm 2 per m2 ≤0.6 cm2 per m2 ≤0.45 cm2 per m2 ≤0.6 cm 2 per m2 Dobutamine stress echocardiography Stress mean gradient NA NA NA NA NA ≥40 mm Hg Stress AVA NA NA NA NA NA ≤1.0 cm2 MDCT Aortic valve
calcification score NA NA • • Men ≥1,200 AUWomen ≥700 AU • • Men ≥2,000 AUWomen ≥1,200 AU NA • • Men ≥2,000 AUWomen ≥1,200 AU
ΔPMean, mean transvalvular gradient; AVA, aortic valve area; AVAi, indexed AVA; BSA, body surface area; MDCT, multidetector CT; NA, not applicable or not available;