Design et synthèse de bicycles peptidiques par réaction
de Ugi-Smiles pour le développement de composés
bioactifs.
Mémoire
Nicolas Paiement
Maîtrise en sciences pharmaceutiques - avec mémoire
Maître ès sciences (M. Sc.)
Design et synthèse de bicycles peptidiques par réaction
de Ugi-Smiles pour le développement de composés
bioactifs
Mémoire
Nicolas Paiement
Maitrise en sciences pharmaceutiques
Maître ès sciences (M. Sc.)
Sous la direction de :
Éric Biron, directeur de recherche
Résumé
Les bicycles peptidiques possèdent des propriétés structurales et pharmacologiques très attrayantes pour le développement de composés bioactifs et sont d’excellents candidats pour la création de chimiothèques visant la découverte d’agents thérapeutiques. Ce type de structure se retrouve dans de nombreux produits naturels démontrant des activités biologiques très variées. Malgré leur grand potentiel, peu d’approches synthétiques efficaces et donnant accès à une grande diversité moléculaire ont été décrites jusqu’à maintenant dans la littérature. Ce mémoire décrit le développement d’une nouvelle approche synthétique sur support solide utilisant la réaction multicomposante de Ugi-Smiles pour préparer des bicycles peptidiques inspirés du Bouvardin et de ses analogues, une famille de produits naturels des plantes Rubiaceae avec des activités anticancéreuses et antimicrobiennes. L’approche présentée donne accès à des systèmes bicycliques complexes et permet l’introduction d’une grande diversité moléculaire. Dans un premier temps, la réaction de Ugi-Smiles a été utilisée pour simultanément ancrer et coupler les deux premiers acides aminés à une résine par leur chaîne latérale. Une étude sur les réactifs et les paramètres a permis d’effectuer les ajustements fonctionnels et structurels sur les composantesqui étaient nécessaires pour une réaction de Ugi-Smiles permettant le clivage de la résine en milieu acide. Par la suite, une première macrocyclisation tête-à-queue suivi de l’élongation du peptide devait permettre une seconde macrocyclisation pour générer un bicycle peptidique pouvant être clivé de la résine. Or, il a été observé que la première cyclisation ne se déroule pas comme prévu et que le système bicyclique désiré n’a pas été obtenu. Une étude a permis d’identifier les éléments problématiques de l’approche et des ajustements sont proposés pour éviter les difficultés de la première macrocyclisation. Malgré tout, des avancements importants ont pu être effectués dans ce projet et l’application des correctifs proposés dans ce mémoire permettra de générer les bicycliques peptidiques ciblés.
Abstract
Peptide bicycles display very attractive structural and pharmacological properties for the development of bioactive compounds and are excellent candidates for the creation of chemical libraries for the discovery of therapeutic agents. This type of structure is found in a great number of natural products with a wide variety of biological activities. Despite their great potential, very few effective synthetic approaches giving access to a large molecular diversity have been described in the literature. This document describes the development of a new synthetic approach on solid support using a Ugi-Smiles multicomponent reaction to prepare peptide bicycles inspired by Bouvardin and its analogs, a family of natural products from Rubiaceae plants with antitumoral and antimicrobial activities. The described approach gives access to complex bicyclic systems and allows the introduction of a large molecular diversity. In a first step, the Ugi-Smiles reaction is used to simultaneously anchor and link the first two amino acids to a resin by their side chain. A study on the reagents and reaction parameters made it possible to carry out functional and structural adjustments on the components allowing the Ugi-Smiles reaction and the cleavage from the resin in an acid medium. Subsequently, a first head-to-tail macrocyclization followed by the elongation of the peptide was supposed to allow a second macrocyclization to generate a peptide bicycle, which can be cleaved from the resin. However, the first cyclization did not go as planned and the desired bicyclic system has not been obtained. A study has identified the problematic elements of the approach and adjustments are proposed to avoid the difficulties of the first macrocyclization. Despite this failure, significant progress has been made in this project and application of the corrections proposed in this document will surely allow the successful synthesis of the targeted bicyclic peptides.
Table des matières
RÉSUMÉ ... II ABSTRACT ... III TABLE DES MATIÈRES ... IV LISTES DES FIGURES ... VI
INTRODUCTION ... VI
CHAPITRE 1 ... VII
CHAPITRE 2 ... VII
CHAPITRE 3 ... IX
CONCLUSION ... X
LISTE DES ABRÉVIATIONS, SIGLES, ACRONYMES ... XI REMERCIEMENTS ... XIV
INTRODUCTION ... 1
1 LES MACROCYCLES PEPTIDIQUES BICYCLIQUES ... 1
2 LA SYNTHÈSE DES MACROCYCLES BICYCLIQUES ... 5
3 LA MACROCYCLISATION PEPTIDIQUE ... 7
4 LA SYNTHÈSE PEPTIDIQUE SUR SUPPORT SOLIDE ET LES MÉTHODES D’ANCRAGE ASSOCIÉES ... 10
5 LES RÉACTIONS MULTICOMPOSANTES ... 14
5.1 La réaction de Ugi-Smiles ... 15
HYPOTHÈSE DE TRAVAIL ET OBJECTIFS ... 19
1 HYPOTHÈSE DE TRAVAIL ... 19
2 APPROCHE ET MÉTHODOLOGIE ... 19
CHAPITRE 1 PREMIER SYSTÈME DE UGI-SMILES ... 24
1.1 PLAN DE SYNTHÈSE ET CARACTÉRISTIQUES DÉSIRÉES DU PRODUIT ... 24
1.2 TESTS PRÉLIMINAIRES DE LA RÉACTION DE UGI-SMILES ... 24
1.3 SYNTHÈSE DES SYNTHONS ... 27
1.3.1 Préparation de l’aldéhyde ... 27
1.3.1.1 Exploration primaire et caractéristiques désirées ... 27
1.3.1.2 Plan de synthèse pour la synthèse de l’ancrage sur résine ... 32
1.3.1.3 Voies synthétiques explorées ... 33
1.3.1.4 Conclusion ... 35
1.3.2 Préparation de l’amine ... 35
1.3.2.1 Plan de synthèse et caractéristiques désirées ... 36
1.3.2.2 Voies synthétiques explorées ... 36
1.3.2.3 Conclusion ... 38
1.3.3 Préparation du nitrophénol ... 38
1.3.3.1 Plan de synthèse et caractéristiques désirées ... 38
1.3.3.2 Voies synthétiques explorées ... 40
1.3.3.3 Conclusion ... 43
1.4 RÉACTION DE UGI-SMILES EN SOLUTION ... 43
CHAPITRE 2 DEUXIÈME SYSTÈME DE UGI-SMILES ... 47
2.1 PLAN DE SYNTHÈSE ET CARACTÉRISTIQUES DÉSIRÉES DU PRODUIT ... 47
2.2 TESTS PRÉLIMINAIRES DE UGI-SMILES ... 48
2.3 SYNTHÈSE DE L’AMINE ... 49
2.3.1 Première hypothèse – p-amidobenzylamine ... 49
2.3.1.1 Voies synthétiques explorées ... 50
2.3.1.2 Vérification de l’aptitude du motif p-amidobenzylamine ... 51
2.3.2 Exploration secondaire ... 53
2.3.2.1 Plan de synthèse de la résine ... 57
2.3.2.2 Voies synthétiques explorées ... 59
2.3.3 Conclusion ... 60
2.4 SYNTHÈSE DE L’ALDÉHYDE ... 61
2.4.1 Plan de synthèse à partir des acides carboxyliques en chaîne latérale ... 62
2.4.1.1 Voies synthétiques explorées ... 65
2.4.1.2 Conclusion ... 71
2.4.2 Plan de synthèse à partir des alcools en chaîne latérale ... 72
2.4.2.1 Voies synthétiques explorées ... 73
2.4.2.2 Conclusion ... 76
2.5 RÉACTION DE UGI-SMILES EN SOLUTION ET EXPORTATION SUR SUPPORT SOLIDE ... 77
CHAPITRE 3 CONSTRUCTION DU BICYCLE PEPTIDIQUE ... 82
3.1 PREMIÈRE MACROCYLISATION ... 82
3.1.1 Tests en solution ... 84
3.1.2 Tests sur support solide ... 87
3.2 SECONDE MACROCYCLISATION ... 89
3.2.1 Tests en solution et sur support solide ... 91
3.3 L’HYPOTHÈSE DE LA RÉACTION DE UGI ... 97
3.3.1 Solution proposée ... 99
CONCLUSION ... 102
ANNEXE GÉNÉRALE ... 105
ANNEXE A – SECTION EXPÉRIMENTALE DU CHAPITRE 1... 106
ANNEXE B – SECTION EXPÉRIMENTALE DU CHAPITRE 2... 110
ANNEXE C – SECTION EXPÉRIMENTALE DU CHAPITRE 3... 117
Listes des figures
Introduction
Figure 1 a) Conformation de la cyclosporine A dans un milieu hydrophobe. b) Conformation de la
cyclosporine A dans un milieu hydrophile ... 1
Figure 2 Comportement des endos et des exopeptidases vis-à-vis un peptide linéaire et un peptide
macrocyclique ... 3
Figure 3 Structure du Bouvardin et des RA I-XVII. Le motif cycloisodityrosine est représenté en bleu et
est entouré de crochets. ... 4
Figure 4 Différentes approches synthétiques répertoriées pour la formation du motif cycloisodityrosine
pour l’étape finale de macrocyclisation. Les réactions nommées en vert ont mené au produit désiré, contrairement à celles nommées en rouge. ... 6
Figure 5 Conditions réactionnelles de l’étape finale de macrocyclisation pour la formation du motif
Aza-cycloisodityrosine. ... 7
Figure 6 Peptide linéaire dans ses configurations trans et cis pour chaque lien peptidique. Les interactions
stériques guidant l’orientation préférentielle sont illustrées par des flèches vertes. La distance entre les extrémités est illustrée par un pointillé noir... 7
Figure 7 Processus de racémisation peptidique par formation d’oxazolone en milieu basique interférant
avec une réaction de couplage peptidique sur support solide. ... 8
Figure 8 Formation de guanidine lors d’une réaction de macrocyclisation avec le HATU comme agent
de couplage. ... 9
Figure 9 Configuration la plus stable de peptides selon l’acide aminé utilisé. La diminution des
interactions stériques par rapport à la configuration opposée est illustrée en vert. ... 9
Figure 10 Mécanisme d’activation d’acide et de couplage peptidique avec une amine pour l’agent de
couplage HATU. ... 12
Figure 11 Mécanisme de formation de DKP lors d’une synthèse peptidique sur support solide en sens
classique et en sens inverse. ... 13
Figure 12 Installation de deux acides aminés sur support solide à l’aide de la méthode d’accrochage BAL.
... 14
Figure 13 Installation de deux acides aminés sur support solide à l’aide la méthode d’accrochage Ugi
analogue à la méthode BAL. ... 14
Figure 14 Mécanisme de la réaction de Ugi. Le carbone chiral formé est identifié par une étoile. ... 15 Figure 15 Réactions de Ugi et de Ugi-Smiles selon leurs principales différences mécanistiques. ... 16 Figure 16 Formation de pont-H stabilisant entre le groupement nitro et l’amine lors du réarrangement
de Smiles. ... 17
Figure 17 Exemple de formation d’un macrocycle peptidique avec une queue lipidique préparé par
réaction de Ugi-Smiles sur support solide. ... 18
Figure 18 Exemple de synthèse avec la méthode envisagée. Toutes les étapes comportant une étoile
impliquent un des degrés d’orthogonalité requis pour les groupements protecteurs... 20
Figure 19 Motif créé après ancrage par méthode BAL, par méthode Ugi et par méthode Ugi-Smiles
Chapitre 1
Figure 1.1 Réaction de Ugi-Smiles effectuée entre la n-butylamine (bleu), le 2-nitrophénol (vert), le
2,4,6-triméthoxybenzaldéhyde (noir) et le tert-butylisonitrile (rose). ... 25
Figure 1.2 Dégradation du produit de Ugi-Smiles avec l'aldéhyde 1 en conditions acides. ... 28
Figure 1.3 Structure des aldéhydes 1-7 utilisés dans les réactions de Ugi-Smiles. ... 29
Figure 1.4 Schéma de préparation d'un ancrage pour support solide avec le motif de l'aldéhyde 6. ... 30
Figure 1.5 Mécanisme de polymérisation du p-aminobenzaldéhyde en poly-imine. ... 32
Figure 1.6 Plan de synthèse vers la résine 10 à partir de l’aldéhyde 8 selon les voies A et B. ... 32
Figure 1.7 Schéma de préparation de résine ChemMatrix® terminée par une fonction acide à l’aide d’un acide aminé espaceur et d’acide succinique. ... 33
Figure 1.8 Ouverture d’anhydride succinique par du aminobenzaldéhyde 9 obtenu par réduction du p-nitrobenzaldéhyde 8. ... 33
Figure 1.9 Couplage peptidique en solution entre le p-aminobenzaldéhyde 9 et l'acide pentanoïque. .. 34
Figure 1.10 Préparation de la résine ChemMatrix 14 terminée par une fonction acide à l'aide d’isoleucine et d'acide glutamique. ... 35
Figure 1.11 Plan de synthèse vers la lysine 17 à partir de la lysine commerciale 15. ... 36
Figure 1.12 Mécanisme de clivage Fmoc intramoléculaire du sel TFA de la Fmoc-Lys-Allyl 17. ... 37
Figure 1.13 Plan de synthèse de la nitro-tyrosine 22 à partir des dérivés commerciaux 18 et 19 selon les voies A, B et C. ... 39
Figure 1.14 Mécanisme d'installation du groupement PhiPr à l'aide de son trichloroacétimidate et du nucléophile à protéger. ... 39
Figure 1.15 Réaction de double protection Alloc sur la nitro-tyrosine et le comportement du produit résultant dans des conditions basiques et de Tsuji-Trost. ... 40
Figure 116 Réaction de protection PhiPr à l'aide de son trichloroacétimidate sur la Fmoc-nTyr-OH. . 41
Figure 1.17 Mécanisme de la formation du trichloroacétimidate du PhiPr. Le NaH peut être utilisé en quantité catalytique puisque l’alcoolate est regénéré au cours de la réaction. ... 43
Figure 1.18 Réaction de Ugi-Smiles entre le p-acétamidobenzaldéhyde (noir), l'Alloc-nTyr-PhiPr 22 (vert), le sel HCl de la Fmoc-Lys-Allyl 17 (bleu) et le tert-butylisonitrile (rose). ... 43
Chapitre 2
Figure 2.1 Produits de Ugi-Smiles et produits de clivages acides résultants pour le premier et pour le second système. ... 47Figure 2.2 Réaction de Ugi-Smiles entre le butyraldéhyde (noir), le pentylisonitrile (rose), le nitrophénol (vert) et la p-méthoxybenzylamine (bleu). ... 49
Figure 2.3 Comportement réactionnel de la p-aminobenzylamine en présence d'un acide activé. ... 50
Figure 2.4 Plan synthétique vers la p-acétamidobenzylamine 30 à partir de p-aminobenzylamine 27. . 50
Figure 2.5 Mécanisme de protection Fmoc sur la p-aminobenzylamine 27 à l'aide de Fmoc-OSu. ... 50
Figure 2.6 Conditions réactionnelles du couplage peptidique entre la Fmoc-p-aminobenzylamine 28 et la résine ChemMatrix terminée par une fonction acide 14. ... 51
Figure 2.7 Réaction de Ugi-Smiles entre la p-acétamidobenzylamine 30 (bleu), le pentylisonitrile (rose), l'Alloc-nTyr-PhiPr 22 (vert) et la Fmoc-AldHoSer-Allyl 45 (noir). ... 52
Figure 2.8 Mécanisme de transamidation soupçonnée de la p-acétamidobenzylamine 30. ... 52 Figure 2.9 Préparation de la résine benzylamine par formation d’amide à partir de résine 14 (a) et par
amination réductive à partir de résine BAL (b). ... 54
Figure 2.10 Réaction d'amination réductive entre le 4-(aminométhyl)benzonitrile 32 et le
2,4,6-triméthoxybenzaldéhyde 1. ... 55 Figure 2.11 Préparation de p-(méthylbutylamido)benzylamine 34 à partir de p-(méthylamino)benzonitrile 32 par couplage peptidique et hydrogénation catalytique. ... 55 Figure 2.12 Clivage par hydrogénolyse du produit de Ugi-Smiles obtenu par condensation de
p-(méthylbutylamido)benzylamine 34 (bleu), 2-nitrophénol (vert), butyraldéhyde (noir) et
pentylisonitrile (rose). ... 56
Figure 2.13 Plan de synthèse vers la résine 36 à partie de la benzylamine 32 ou de la benzylamine 35
selon les voies A, B et C. ... 58
Figure 2.14 Préparation de la résine 36 à partir de la benzylamine 35 et de la résine ChemMatrix®
terminée par une fonction acide 14, puis son couplage à une Fmoc-Gly-OH pour permettre
un dosage du groupement Fmoc. ... 59
Figure 2.15 Réaction de déprotonation d'un acide carboxylique par du DiBAl. ... 63 Figure 2.16 Mécanisme de réduction d'un amide et d'un amide de Weinreb par le DiBAl. Les voies
mécanistiques menant à l'amine (bleu) et à l'aldéhyde (rouge) sont illustrées. La forme cyclique stabilisatrice adoptée par l'amide de Wienreb favorisant l’obtention de l’aldéhyde est aussi représentée. ... 64
Figure 2.17 Différentes voies de synthèse envisagées pour la synthèse des aldéhydes 45 et 46 à partir des
esters 41 et 42. Acide aspartique (n = 1), Acide glutamique (n = 2). Réactions : i) Protection
Allyl. ii) Clivage tBu. iii) Réduction en alcool. iv) Couplage peptidique vers l’amide de Weinreb. ... 65
Figure 2.18 Réduction de l'ester 45 à l'aide de DiBAl. Les produits observés sont indiqués avec la
proportion finale obtenue et les équivalents de DiBAl nécessaires à leur formation. ... 66
Figure 2.19 Synthèse du Fmoc-Asp-OAllyl 53 à partir de l'ester 41. L’équivalent Glu de chaque produit
illustré porte le numéro n+1. ... 67
Figure 2.20 Réduction de l'acide 53 en alcool 47, à l'aide de méthylchloroformate et de NaBH4. L’équivalent Glu de chaque produit illustré porte le numéro n+1. ... 67
Figure 2.21 Équilibre entre la forme linéaire et cyclique des alcools des acides glutamique et aspartique 47 et 48 par transestérification intramoléculaire. ... 68 Figure 2.22 Préparation de l'amine de Wienreb 50 par couplage peptidique de l'acide 54 et du produit
résultant de la tentative subséquente de remplacement du Fmoc par un Boc. ... 69
Figure 2.23 Plan synthétique vers l'aldéhyde 46 à partir de l'ester 42 par protection benzyl totale de
l'amine et de l'acide α. L’équivalent Asp de chaque produit illustré porte le numéro n-1. . 70
Figure 2.24 Produits obtenus par protection benzyl d'acide glutamique γ méthyl ester. ... 70 Figure 2.25 Synthèse de l'alcool d'acide aspartique tribenzylé 65 à partir de son ester tBu 57 par réduction
de l’acide correspondant. L’équivalent Glu de chaque produit illustré porte le numéro n+1. ... 71
Figure 2.26 Oxydation de Dess-Martin de l’alcool 65 vers l’aldéhyde 59. L’équivalent Glu de chaque
produit illustré porte le numéro n+1. ... 71
Figure 2.27 Mécanisme de formation de lactone d'Homosérine 55 lorsque l'acide est protégé Allyl et
Figure 2.28 Préparation de Fmoc-Ser-Allyl 69 à partir de Fmoc-Ser(OtBu)-OH 67. L'attaque nucléophile
résultant en une formation potentielle de lactone est montrée en rouge. ... 73
Figure 2.29 Mécanisme de clivage du Fmoc lors de l'oxydation de Swern de la Fmoc-Ser-Allyl 69. Les
pKa indiqués correspondent à ceux des protons indiqués en rouge. ... 74
Figure 2.30 Mécanisme du clivage du groupement protecteur trityl de la Fmoc-HoSer-OH 71 en milieu
acide, résultant en la formation de lactone 55. ... 74 Figure 2.31 Mécanisme de transestérification de l'homosérine 47 vers la formation de lactone 55 de la
Fmoc-HoSer-Allyl en milieu neutre. ... 76
Figure 2.32 Préparation de Fmoc-AldHoSer-Allyl 45 à partir de H2N-HoSer-OH 72. ... 76 Figure 2.33 Voie synthétique inefficace pour l'obtention de AldGlu-Allyl 46 à partir de
Fmoc-Glu-Allyl 54 dû à la réaction infructueuse de réduction de l'acide en alcool. ... 77 Figure 2.34 Réaction de Ugi-Smiles entre le butyraldéhyde (noir), le 2-nitrophénol (vert), le
pentylisonitrile et la benzylamine ou la p-(méthylbutylamido)benzylamine 38 (bleu). ... 78 Figure 2.35 Réaction de Ugi-Smiles entre la benzylamine (bleu), le pentylisonitrile (rose), la
Fmoc-AldHoSer-Allyl 45 (noir) et l'Alloc-nTyr-PhiPr 22 (vert). ... 78 Figure 2.36 a) Produit de Ugi-Smiles obtenu lors du couplage de la benzylamine (bleu), le pentylisonitrile
(rose), la Fmoc-AldHoSer-Allyl 45 (noir) et l'Alloc-nTyr-PhiPr 22 (vert), mais avec le
groupement PhiPr clivé. b) Produit de Ugi résultant du couplage des mêmes réactifs, mais où la nito-tyrosine a réagi par l'acide libre. La masse de l’ion moléculaire attendue lors d’une analyse LCMS-ESI+ est aussi indiquée pour chaque produit. L’étoile représente un carbone chiral en proportions racémiques. ... 79
Figure 2.37 Produit de Ugi-Smiles obtenu par condensation de la résine 36, le pentylisonitrile (rose), la
Fmoc-AldHoSer-Allyl 45 (noir) et l'Alloc-nTyr-PhiPr 22 (vert), ainsi que le produit de
Passerini obtenu avec la résine acide 14 résiduelle et les mêmes réactifs libres. ... 80 Figure 2.38 Résine 74 terminée par la fonction p-(méthylamido)benzylamine (bleu), mais précédé d'un
ancrage Rink amide (vert) et d'un acide glutamique (rose). Le site de clivage dans un milieu acide est indiqué par une vague noire. ... 80
Chapitre 3
Figure 3.1 Formation du premier macrocycle peptidique par couplage peptidique de l’acide et de l’amine
protégés Allyl et Alloc respectivement. Le lien peptidique et ses composantes sont encerclés en orange... 82
Figure 3.2 a) Illustration d'une potentielle impossibilité de cyclisation pour le petit cycle dû à un
éloignement trop important des extrémités à cycliser. b) Représentation de l’inversion des étapes de macrocyclisation, où les extrémités du petit cycle pourraient être plus rapprochées. ... 83
Figure 3.3 a) Produit de Ugi-Smiles entre la benzylamine (bleu), le pentylisonitrile (rose), la
Fmoc-AldHoSer-Allyl 49 (noir) et l'Alloc-nTyr-PhiPr 22 (vert). b) Produit de Ugi-Smiles entre la
résine Rink-benzylamine 74 (bleu), le pentylisonitrile (rose), la Fmoc-AldHoSer-Allyl 45 (noir)
et l'Alloc-nTyr-PhiPr 22 (vert). ... 84 Figure 3.4 Mécanisme de capture de l'allyl par l'amine lors du clivage Allyl/Alloc par réaction de
Figure 3.5 Mécanisme du test au chloranil pour révéler la présence d'amines primaires et secondaires. La
couleur du produit et du réactif est indiquée. ... 88
Figure 3.6 Plan de synthèse sur support solide du bicycle sans élongation de la chaîne peptidique à partir
du produit de première cyclisation. ... 89
Figure 3.7 Comparaison entre le Bouvardin et son analogue le plus semblable pouvant être obtenu avec
la méthode développée, encore accroché à la résine. ... 90
Figure 3.8 Peptide linéaire obtenu après l'élongation du premier macrocycle de séquence H 2N-Ala-Ala-Tyr-Ala-AldHoSer-nTyr-PhiPr. ... 92
Figure 3.9 Différentes strucutres de même masse moléculaire pouvant correspondre au produit observé
lors d'une analyse LCMS. a) Produit bicyclique auquel l'oxyde de phosphine est lié de manière électrostatique. b) Produit linéaire auquel la phosphine est liée de manière covalente pour former l’ester activé phosphonium. c) Produit linéaire auquel la phosphine est liée de manière covalente pour former l’imine de phosphore. ... 94
Figure 3.10 Mécanisme d'activation d'un acide carboxylique par le PyAOP en présence de DiPEA. Les
divers intermédiaires et produits réactionnels sont identifiés. ... 95
Figure 3.11 Structure probable du produit linéaire obtenu après tentative de macrocyclisation et couplage
d'une Fmoc-Ala-OH, selon la masse moléculaire observée lors d'une analyse LCMS du produit clivé. ... 96
Figure 3.12 Réaction du produit linéaire après une tentative de cyclisation avec une solution de pipéridine
et la structure des divers produits obtenus. ... 97
Figure 3.13 Produit de Ugi obtenu après condensation des produits de départ sur la résine benzylamine
et son produit de macrocyclisation. ... 98
Figure 3.14 Voies mécanistiques probables de l'hydrolyse de l'éther de phosphonium en présence de
pipéridine dans le DMF. ... 99
Figure 3.15 Plan de synthèse proposé à partir d'une protection méthyle au lieu de PhiPr sur l'acide de la
nitro-tyrosine. ... 101
Conclusion
Figure 1 Préparation de nitro-tyrosine Alloc-nTyr-Et comme remplacement stable de la nitrotyrosine
Alloc-nTyr-PhiPr 22. ... 103 Figure 2 Voies synthétiques inexplorées. (a) Oxydation de l’homosérine 47 au DMP. (b) Réduction au
DiBAl du Bn2-Asp(OBn)-Bn. (c) Réduction du nitrile en benzylamine 34 par LiAlH4 ou par Nickel de Raney. (d) Couplage peptidique de la benzylamine 39 sur la résine acide 14. ... 104
Liste des abréviations, sigles, acronymes
°C Degrés Celsius
Å Angstrom
Ac Acétyl
AcOEt Acétate d’éthyle
Ala L-Alanine
AldGlu L-Acide 2-amino-5-oxopentanoique AldHoSer L-Acide 2-amino-4-oxobutanoique
Alloc Allyloxycarbamate
APCI+ Ionisation chimique positive à pression atmosphérique
Asp L-Acide aspartique
atm Pression atmosphérique
BAL Backbone amide linker
Bn Benzyl
Boc tert-Butyloxycarbamate
BOP Benzotriazol-1-yloxytris(diméthylamino)phosphonium hexafluorophosphate
CCM Chromatographie sur couche mince
conc Concentré
DBU Dibenzofulvène
DCM Dichlorométhane
DiBAl Hydrure de Diisobutylaluminium
DiPEA N,N-Diisopropyléthylamine DKP Dicétopipérazine DMAP 4-Diméthylaminopyridine DMF N,N-Diméthylformamide DMP Périodinane de Dess-Martin DMSO Diméthylsulfoxyde DNPH 2,4-Dinitrophénylhydrazine EM Molarité effective eq Équivalent
ESI+ Ionisation positive par électronébuliseur
Et Éthyl
FDA Food and Drug Administration
Fmoc Fluorénylméthoxycarbonyle
GCPR Récepteur couplé aux protéines G
Glu L-Acide glutamique
HATU Hexafluorophosphate de-N,N-diméthylméthaniminium
HFIP Hexafluoroisopropanol
HOAT 1-Hydroxy-7-azabenzotriazole
HOBT Hydroxybenzotriazole
HoSer L-Homosérine
HPLC Chromatographie en phase liquide à haute performance
Hz Hertz
IC50 Concentration inhibitrice médiane
Ile Isoleucine
J Constante de couplage
LCMS Chromatographie en phase liquide-spectrométrie de masse
Leu L-Leucine
Lys L-Lysine
M Concentration (mole/litre)
m Masse
m/z Ratio masse sur charge
M+H Masse de l'ion moléculaire avec un proton
MCR Réaction multi-composantes Me Méthyl MHz Mega Hertz min Minute mol Mole MS Spéctrométrie de masse MW Micro-ondes NMM N-Méthylmorpholine nTyr L-3-nitrotyrosine o Ortho o-NBS ortho-nitrobenzènesulfonyl OSu Hydroxysuccinimide p Para Pbf 2,2,4,6,7-Pentaméthyldihydrobenzofuran-5-sulfonyl PCC Pyridinium chlorochromate
Pd/C Palladium sur charbon
PDC Pyridinium dichromate
PEG Polyéthylène glycol
PFP Pentafluorophényle
PG Groupement protecteur
pH Potentiel hydrogène
PhiPr 2-Phenylisopropyl
pKa Constante de dissociation acide PPI Interactions protéines-protéines
ppm Partie par million
Pro L-Proline
TSA/
p-TsOH Acide para-toluènesulfonique
PyAOP (7-Azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate
rdt Rendement
Rink 4-[(R,S)-(2,4-diméthoxyphenyl)(Fmoc-amino)méthyl]phenoxyacetic acid
RMN Résonance magnétique nucléaire
RP-HPLC Chromatographie en phase liquide à haute performance en phase inverse
sat Saturé
sec Seconde
Ser L-Sérine
SN1 Substitution nucléophile monomoléculaire SNAr Substitution nucléophile aromatique SPPS Synthèse peptidique sur support solide
tBu tert-Butyl
TFA Acide trifluoroacétique
THF Tetrahydrofurane
TIPS Triisopropylsilane
TLC Chromatographie sur couche mince
tR Temps de rétention
Trt Trityl
TTN Nitrate de thallium(III) trihydraté
Tyr L-Tyrosine
Remerciements
Pour commencer, je veux adresser mes remerciements à mon directeur de maîtrise, le Pr Éric Biron, pour sa grande disponibilité, ses encouragements et ses conseils tout au long de mon projet et de la rédaction de ce mémoire.
Je remercie également tous les membres du groupe de recherche pour leur aide précieuse, leur écoute et leur support, particulièrement Noémie Régnier et Louis-David Guay.
Je remercie enfin les membres de ma famille qui ont été à mes côtés pendant ce projet et qui m'ont toujours encouragé, sans oublier bien sûr mes proches et mes amis qui m’ont soutenu pendant ce long processus.
Introduction
1 Les macrocycles peptidiques bicycliques
Le domaine pharmaceutique est en constante recherche de nouveaux composés d’intérêt pour le développement de médicaments, et ce depuis la découverte de plantes médicinales. Avec les années, l’expérience de l’humanité en la matière ainsi que le développement de méthodes d’analyse et de purification ont permis d’établir certains principes d’efficacité pour les principes actifs. Ainsi, en 1997, Chrisopher A Lipinski a fait ressortir cinq tendances que partagent les médicaments biodisponibles oralement, désormais connue comme la règle des 5. Pour qu’un médicament soit suffisamment soluble et perméable, il doit donc avoir au plus 5 donneurs et 10 accepteurs de pont H, la masse moléculaire doit être plus petite que 500 g/mol et le coefficient de partage octanol/eau (LogP) ne doit pas dépasser 5.1 Une violation de plus de l’un de ces critères devrait
donc rendre une molécule d’intérêt indisponible oralement.
Pourtant, il existe de nombreuses exceptions à cette règle, dont beaucoup de composés macrocycliques peptidiques.2, 3 Ce sont de petits polymères d’acides aminés comportant un cycle d’au moins 12 atomes et de
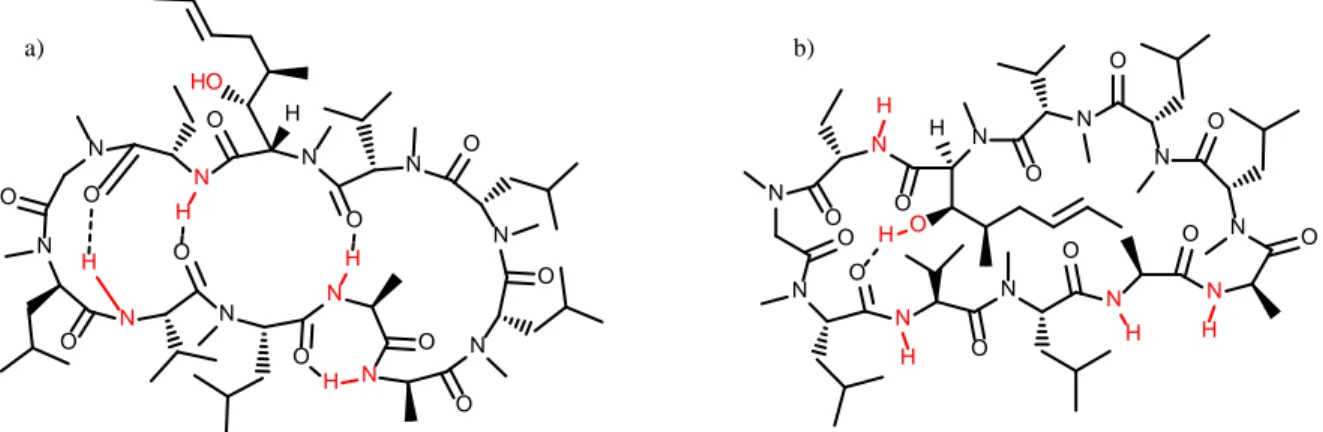
masse moléculaire généralement comprise entre 500 et 2000 Dalton. L’un des meilleurs exemples pour illustrer ce phénomène est le cas de la cyclosporine A qui, malgré sa masse moléculaire de 1202,6 g/mol et ses 5 donneurs et 12 accepteurs de ponts H, présente une perméabilité membranaire et une biodisponibilité surprenante. Ce comportement peut être expliqué par sa structure macrocyclique qui lui permet de cacher ses fonctions polaires et ses ponts H vers l’intérieur dans les milieux lipophiles, à l’instar des protéines qui développent une structure secondaire relative à leur environnement (Figure 1).3-5
N N N N N N N N N N N O O O O O O O O O O HO H H O H H H N N N N N N N N N N N O O O O O O O O O O O O H H H H H H a) b)
Figure 1 a) Conformation de la cyclosporine A dans un milieu hydrophobe. b) Conformation de la cyclosporine A dans un milieu hydrophile.
Cette propriété de la structure macrocyclique à mimer une structure secondaire est l’un des éléments clés qui la rend si intéressante au niveau pharmaceutique et pourquoi elle est de plus en plus explorée en recherche.5
pharmacodynamiques. La rigidité du cycle permet une pré-organisation structurelle, donc l’entropie de liaison avec la cible thérapeutique est diminuée, puisque le macrocycle n’est pas constamment en train de tourner et est donc immédiatement dans la bonne configuration.4, 5 La conséquence de cette caractéristique est double;
d’une part la force de liaison avec la cible est augmentée et de l’autre le médicament est plus sélectif envers la cible désirée.
Un bon exemple de ce phénomène est une expérience menée par Kim, Y.-K. et al. où, sur une chimiothèque de 122 molécules macrocycliques et leur analogue linéaire, seuls 2 macrocycles ont répondu positivement à plus d’un des 40 tests cellulaires divers effectués, contre 20 linéaires.6 Aussi, beaucoup moins de macrocycles
ont eu une interaction lors d’au moins l’un des tests, soit 19 contre 33, renforçant encore une fois la conclusion de leur sélectivité augmentée. Quant à l’interaction avec la cible, il a été démontré que la forme macrocyclique d’un inhibiteur particulier de la farnesyltransférase a une IC50 de 0,1 nM, comparativement à 5490 nM pour son analogue linéaire.7
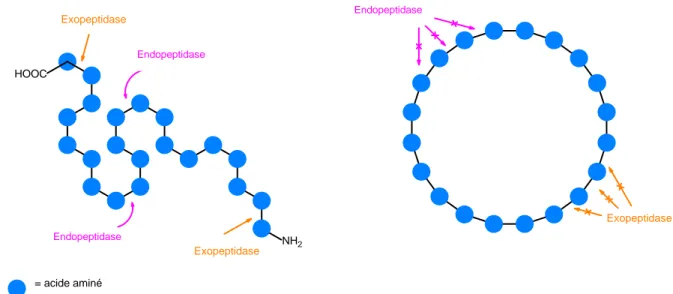
Plus spécifiquement, les macrocycles peptidiques démontrent plusieurs avantages au niveau pharmacocinétique par rapport à leur analogue linéaire. En effet, en plus de leur biodisponibilité très faible, la durée de vie des peptides linéaires est très courte en milieu biologique, car de nombreuses protéases les métabolisent, les rendant ainsi quasi inutilisables comme médicaments malgré leur grande activité. Les peptides macrocycliques, quant à eux, sont beaucoup plus difficiles à métaboliser par les exo- et endopeptidases, puisqu’ils n’adoptent pas nécessairement la configuration feuillet β dont elles ont besoin pour reconnaître et hydrolyser les liaisons peptidiques (Figure 2).3, 8 En d’autres mots, la sélectivité augmentée des macrocycles
peptidiques par leur rigidité conformationnelle s’applique aussi aux protéases, puisqu’elle empêche la reconnaissance pour la protéolyse. De plus, la cyclisation d’un peptide réduit son hydrophilicité car le lien créé, que ce soit amide, disulfure ou autre, est généralement plus apolaire que ses composantes. Enfin, contrairement aux petites molécules, plus couramment étudiées en recherche pharmaceutique, les métabolites des peptides sont connus comme étant non toxiques et sans risques de bioaccumulation.2, 9, 10
HOOC NH2 Exopeptidase Exopeptidase Exopeptidase Exopeptidase Exopeptidase Exopeptidase = acide aminé
Figure 2 Comportement des endopeptidases et des exopeptidases vis-à-vis un peptide linéaire et un peptide macrocyclique.
Dû à leurs propriétés si différentes des petites molécules, il est logique que lesapplications des peptides macrocycliques soient aussi différentes, puisque pour des cibles plus complexes, on ait besoin d’agents thérapeutiques plus complexes.10 En effet, les peptides macrocycliques ouvrent la porte à de nouveaux types
d’interactions généralement inaccessibles aux petites molécules, par exemple la modulation des interactions protéines-protéines (PPI) ou le blocage des récepteurs reconnaissant des domaines ou des motifs discontinus. Les champs où ils sont le plus étudiés sont donc dans leurs interactions avec les protéases, dans les récepteurs couplés aux protéines G (RCPG) et dans la disruption des interactions entre les protéines d’un milieu.8 Ces
interactions spécifiques peuvent se traduire par diverses activités macroscopiques, comme un effet anticancéreux, anti-inflammatoire, antibactérien ou antifongique, ce qui est très recherché présentement dans l’industrie pharmaceutique.11 De plus, comme seulement 10% des RCPG sont présentement ciblés par des
médicaments sur le marché, il y a une grande opportunité pour les macrocycles peptidiques.12 Ainsi, plus d’une
centaine de peptides ont été approuvés par la FDA et sont actuellement disponibles sur le marché.13
Parmi ces peptides macrocycliques, quelques cas phares valent la peine d’être discutés. La vancomycine par exemple, est un puissant antibiotique développé dans les années 50 qui agit sur les bactéries Gram-positif, principalement sur Staphylococcus aureus, par l’inhibition de la formation de peptidoglycanes pour la paroi cellulaire.14,15 Malgré ses multiples inconvénients, allant de la biodisponibilité orale quasi nulle à une gamme
d’effets secondaires importants, ce macrocycle peptidique est encore utilisé aujourd’hui grâce à sa grande efficacité. L’ocytocine, la vasopressine et l’insuline, trois autres peptides d’origine naturelle, sont aussi utilisés comme agents thérapeutiques, en plus de leur action naturelle chez l’homme, respectivement pour réduire les saignements après l’accouchement,10 comme antidiurétique16 et pour le contrôle du glucose sanguin.17
Endopeptidase
Endopeptidase
Enfin, pour quelques exemples de peptides macrocycliques non commerciaux mais tout de même intéressants, STFI-1, un inhibiteur de la trypsine trouvé dans les tournesols, a été étudié dans l’optique de développer des peptides avec une activité angiogénique grâce à sa forte affinité avec sa cible (IC50 0,002 nM).18 Le Bouvardin
et sa famille de molécules, les RA I-XVII, sont quant à eux des alcaloïdes bicycliques comportant un motif cycloisodityrosine responsable de leur effet antitumoral prometteur (Figure 3).11, 19 Ainsi, de nombreux analogues
ont été préparés et leurs propriétés ont été extensivement étudiées. L’un des analogues les plus prometteurs, le RA-V, montre par exemple un effet inhibiteur de la croissance cellulaire à une concentration de 60 nM dans les cellules KB, une lignée de cellules tumorales.20 Les avantages des peptides bicycliques sont les mêmes que
ceux des peptides simplement macrocycliques, mais accentués, puisqu’ils sont plus rigides, plus stables et plus spécifiques. O N R1O N HN NH N NH O O O O O O OR2 R3 R6 R5 Tyr Tyr Ala Tyr Ala Ala R4
Figure 3 Structure du Bouvardin et des RA I-XVII. Le motif cycloisodityrosine est représenté en bleu et est entouré de crochets.
Le Bouvardin et ses analogues naturels comme synthétiques ont été étudiés pendant plus de 40 ans pour leur activité anticancéreuse contre plusieurs lignées de cellules cancéreuses et leur activité antimicrobienne sur plusieurs souches bactériennes.21 Leur mécanisme d’action a été amplement étudié et semble provenir de leur
liaison à l’unité ribosomale 80S, résultant en une inhibition de la synthèse protéique.11, 19 Les membres naturels
de cette famille de bicycles peptidiques ont été isolés de Rubia akane ou d’autres rubiacées, d’où leur nom, les RA.22 Parmi la multitude d’analogues naturels, le RA-VII et le Bouvardin ont la meilleure activité et, au vu de la
quantité d’analogues synthétiques et du volume d’études qui leur a été consacré, cette classe de molécule a définitivement un potentiel intéressant au niveau pharmaceutique.23, 24 Une revue des analogues a d’ailleurs
démontré que le pharmacophore est leur motif cycloisodityrosine et que le reste du peptide sert principalement à rigidifier et à moduler la structure pour mener à la bonne pré-organisation structurelle.11, 24 La plupart des
hydrosolubilité, sa perméation membranaire limitée ou pour optimiser sa sélectivité envers sa cible, mais peu d’exploration à large spectre a été effectuée pour diversifier l’activité de ses analogues.25, 26
Une grande majorité des médicaments discutés sont des produits naturels et non des analogues synthétiques ou des produits de rationalisation. Ainsi, malgré leur potentiel thérapeutique important, le domaine pharmaceutique semble encore timide à l’exploitation des macrocycles peptidiques. La principale raison pour cette incohérence est la difficulté synthétique importante qui, principalement pour les cycles de 3, 4 ou 5 acides aminés, peut être un réel défi.27
2 La synthèse des macrocycles bicycliques
Pour préparer le Bouvardin et ses analogues, diverses méthodes ont été explorées. Bien sûr, puisque la partie complexe de ces molécules au niveau synthétique est leur motif cycloisodityrosine, c’est sur sa préparation que la majeure partie des études ont été menées. Ainsi, pour fermer ce cycle à 14 atomes fortement tendu par la présence des deux groupements aromatiques, la plupart des méthodes tentées se sont révélées infructueuses. Parmi celles-ci, les plus notables sont la macrolactamisation, le couplage catalytique de Ullmann entre le C3 et le O2 et un couplage oxydatif classique du phénol.11 Plusieurs autres méthodes ont en revanche réussi à divers
degrés à mener au produit désiré, soient un couplage oxydatif promu par du trinitrate de thallium tri-hydraté (TTN),28 un couplage de Ullmann entre le C1 et le O229 et, plus notablement, les deux SNAr possibles (Figure
O N R3O O N MeO HN O O MeO Cbz OH N MeO O O MeO OH N O2N N O O MeO Boc F OH N HO HN O O MeO R1 R2 I I OH O NH OMe O HO Br BrCl Cl OH N HO HN O O MeO Cbz Br BrCl Cl TTN F N HO N O O MeO Boc OH NO2 SNAr SNAr 1 2 3 Ullmann Ullmann lactamisation Couplage oxydatif Meilleur rendement R4 R5
Figure 4 Différentes approches synthétiques répertoriées pour la formation du motif cycloisodityrosine pour l’étape finale de macrocyclisation. Les réactions nommées en vert ont mené au produit désiré, contrairement à celles nommées en rouge.
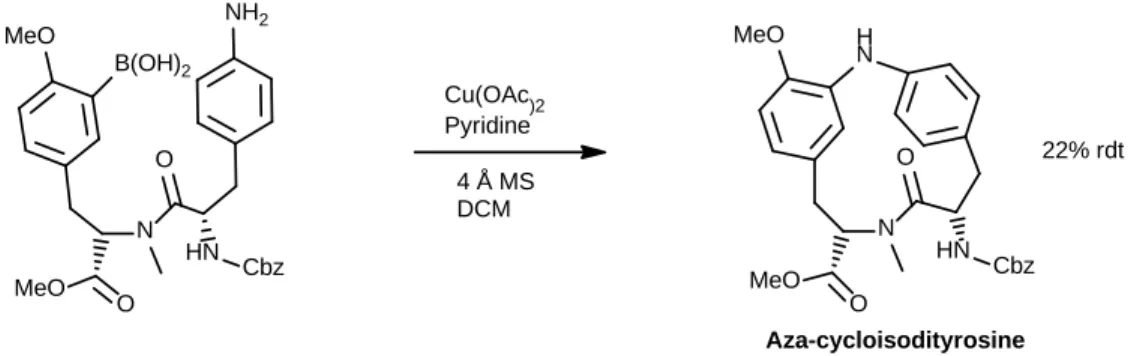
L’une des modifications structurelles les plus pertinentes effectuée sur le RA-VII, fût de substituer l’oxygène du lien biaryl éther par un azote pour vérifier son impact sur le pharmacophore. Ce composé aza-cycloisodityrosine fût préparé par couplage assisté par de l’acétate de cuivre entre l’aniline et l’acide boronique du produit linéaire pour obtenir le macrocycle dans un faible rendement (Figure 5). Il a ensuite été démontré que ce produit est 5 à 7 fois moins cytotoxique que le RA-VII sur les souches cancéreuses HL-60 et HTC-116, respectivement de leucémie et du cancer du côlon.32 Il semble donc que l’aniline, plus riche en électrons que l’éther, réduit la force
de liaison avec l’unité ribosomale 80S de ces cellules. Cependant, peu d’autres modifications significatives ont été effectuées au niveau du pharmacophore pour en déterminer l’effet.
H N N MeO HN O O MeO Cbz NH2 N MeO HN O O MeO Cbz B(OH)2 Cu(OAc)2 Pyridine 4 Å MS DCM 22% rdt Aza-cycloisodityrosine
Figure 5 Conditions réactionnelles de l’étape finale de macrocyclisation pour la formation du motif aza-cycloisodityrosine.
3 La macrocyclisation peptidique
Les macrocyclisations peptidiques sont reconnues pour être difficiles et traitables presque exclusivement au cas par cas sans possibilité d’approche universelle.33 La raison de ce défi synthétique est l’élévation de l’énergie de
l’état de transition lors du rapprochement des extrémités. En effet, les liens peptidiques adoptent, au niveau énergétique le plus bas, une conformation trans, se traduisant par une linéarité favorisée de la chaîne.34 Une
rotation de ces liens augmente les interactions défavorables entre les chaînes latérales, donc le peptide n’a pas tendance à adopter la configuration requise pour cycliser (Figure 6).35, 36 De plus, ces macrocycles doivent
adopter des angles et des longueurs de liaisons défavorables comparativement à leur forme linéaire pour adopter le bon état de transition, résultant en une tension de cycle importante.37, 38 Cet effet est particulièrement
marqué pour les cycles de 8 à 13 atomes, alors qu’au-dessus de 21, le cycle peut tolérer une exclusivité de liens trans puisque les interactions de chaînes latérales y sont moins importantes.27 En effet, plus la taille du
cycle est grande, plus les angles de liaisons approchent d’une linéarité apparente.
H N N H NH O O O H2N R1 R2 R3 R4 O OH Trans Grande distance H N HN NH O O O NH2 R1 R2 R3 R4 O HO Faible distance cis cis cis
Plus stable Moins stable
Trans Trans
Figure 6 Peptide linéaire dans ses configurations trans et cis pour chaque lien peptidique. Les interactions stériques guidant l’orientation préférentielle sont illustrées par des flèches vertes. La distance entre les extrémités est illustrée par un pointillé noir.
En termes plus pratiques, cette réactivité diminuée augmente significativement les temps de réactions de cyclisation comparativement aux mêmes réactions de couplage sur des analogues linéaires. Le principal effet observé de ce phénomène est une oligomérisation favorisée, puisque la réaction intermoléculaire est plus rapide
que son équivalent intramoléculaire et est donc favorisée. Les systèmes sont alors définis par leur molarité effective (EM), soit le ratio des vitesses de formation intra et intermoléculaires dans l’optique de comparer les systèmes et de définir la facilité de cyclisation relative.39 Aussi, bien que l’utilisation d’agents de couplage pour
la formation de liens amides soit très répandue et généralement reconnue comme étant la meilleure approche,40
des contraintes supplémentaires apparaissent lorsqu’il est question de macrocyclisations. En effet, la réactivité augmentée des esters activés formés par les agents de couplage offre une possibilité de racémisation plus ou moins importante du carbone α de l’acide aminé selon l’agent utilisé (Figure 7).41, 42 Bien que l’épimérisation soit
une considération importante lors de n’importe quel couplage peptidique, car la stéréochimie des acides aminés est cruciale pour la reconnaissance biologique et l’activité des peptides, il est d’autant plus important de contrôler ce paramètre lors d’une macrocyclisation, car la durée de vie prolongée de l’ester activé avant sa substitution par l’amine augmente les risques de participation à l’équilibre menant à une racémisation.43, 44
H N N H O R2 R1 O X HN R2 N O O R Base H N R2 N O O R Base Base H N R2 N O O R oxazolone oxazolone H N N H O R2 R1 O NHR3 NH2R3 NH2R3 H N N H O R2 R1 O NHR3 Racémisation Lent Vitesse variable
Figure 7 Processus de racémisation peptidique par formation d’oxazolone en milieu basique interférant avec une réaction de couplage peptidique sur support solide.
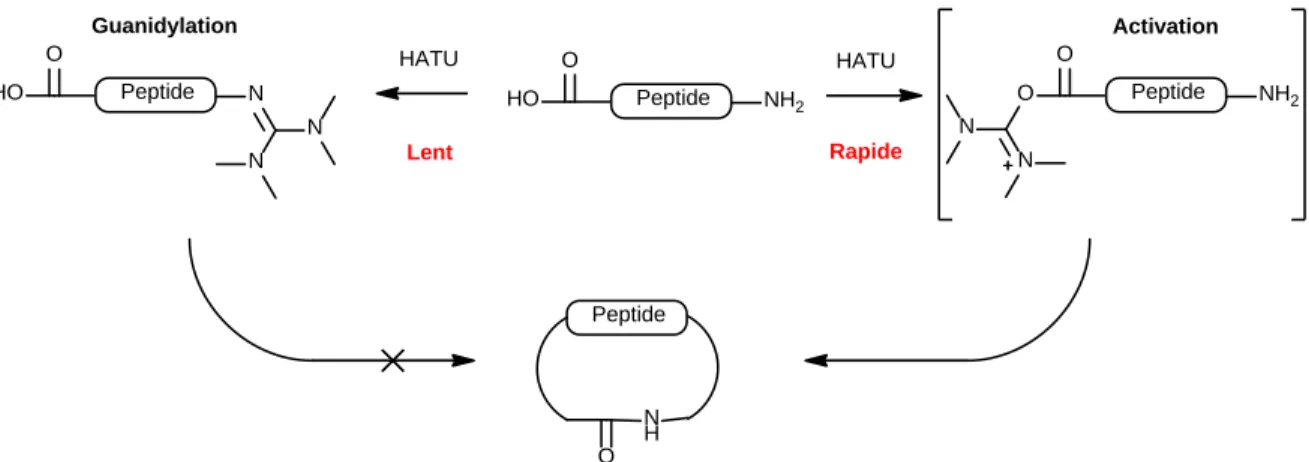
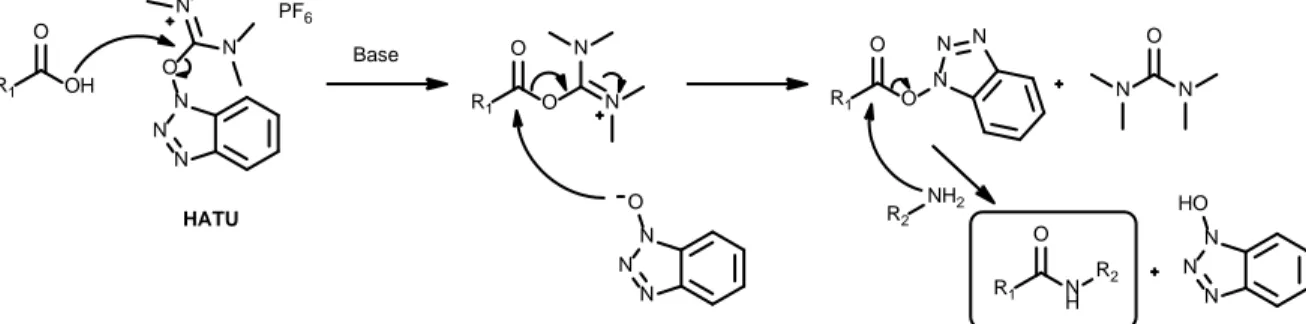
Les agents de couplage à base d’uronium comme le HATU, dont l’efficacité est avérée pour les couplages peptidiques classiques, ne sont pourtant pas recommandés lors de macrocyclisations peptidiques lentes. En effet, lorsque mis en présence d’amines sans acides, ces agents de couplages forment une guanidine à l’extrémité amine, l’empêchant ainsi de réagir (Figure 8).45 Puisque les fonctions acides et amines ne se
rencontrent pas souvent lors d’une macrocyclisation difficile, l’amine peut alors être bloquée avant de rencontrer l’ester activé et ainsi diminuer fortement les rendements. La meilleure solution à ce problème spécifique est l’utilisation de dérivés phosphoniums, comme le PyAOP et tous les autres analogues du BOP, qui ne peuvent créer de guanidine et donc permettent une réaction sans risque de blocage peu importe la durée.46, 47
Peptide N O HO Peptide NH 2 O HO Peptide NH2 O O N N N N Peptide N H O HATU HATU Lent Rapide Activation Guanidylation
Figure 8 Formation de guanidine lors d’une réaction de macrocyclisation avec le HATU comme agent de couplage.
Vu la variabilité des réactions de macrocyclisation, de nombreuses études ont été menées pour rationaliser le processus et fournir différentes méthodes pour optimiser les chances de réussite. Premièrement, comme mentionné plus tôt, la linéarité du peptide est dictée par l’encombrement des chaînes latérales, donc la conformation trans est moins énergétique. Cependant, il existe plusieurs situations où l’orientation cis est plus stable et donc où la rotation du peptide vers une conformation plus près de l’état de transition recherché est atteinte.27 Ainsi la proline, le seul acide aminé naturel ayant une amine secondaire et non primaire, force une
rotation dans la chaîne peptidique par la présence de son noyau cyclopentane, rapprochant de ce fait les extrémités. Une N-méthylation et la présence d’acides aminés D ont le même effet puisque, dans chaque cas, l’encombrement stérique apporté par le groupement sur l’azote ou par la distance entre les chaînes latérales inverse la configuration la plus stable (Figure 9).48-50
H N N H NH O O O H2N R1 R2 R3 R4 O OH N N H NH O O O H2N R1 R2 R4 O OH H N N H NH O O O H2N R1 R2 R4 O OH R3 N NH N H O O O H2N R1 R2 R4 O OH R3 Peptide trans
Acide aminé D Proline N-Méthylation
Figure 9 Configuration la plus stable de peptides selon l’acide aminé utilisé. La diminution des interactions stériques par rapport à la configuration opposée est illustrée en vert.
La quantité et la position de ces agents de rotation dans la chaîne à cycliser ont une grande importance sur le résultat obtenu. En effet, pour former le peptide cyclique cyclo[Pro‐Ala‐Ala‐Phe‐Leu], seul le couplage final
entre l’alanine et la phénylalanine a mené à l’obtention du produit par le groupe de Schmidt, et ce avec seulement 21% de rendement.51 Les tentatives de cyclisation aux autres positions ont mené à une majorité de
dimère et même à un rendement nul avec la proline comme acide aminé terminal, car son encombrement stérique trop important nuit davantage au couplage. Ainsi, la présence d’une proline ou de n’importe quel autre agent de rotation n’est pas suffisant, sa position dans le cycle est aussi cruciale et doit mener à un maximum de rapprochement des extrémités avec un minimum d’encombrement stérique.52, 53 De plus, la présence de
plusieurs agents de rotation donne généralement de meilleurs résultats, sauf dans le cas où tous les acides aminés sont D. Dans ce cas, aucune rotation n’est induite, puisque c’est l’alternance de stéréochimie inverse qui induit le changement d’orientation.54, 55
Enfin, la plus importante considération pour discriminer entre le couplage intra- et intermoléculaire est le facteur de dilution. En effet, lorsque le peptide est fortement dilué, la vitesse de réaction intermoléculaire est diminuée proportionnellement par la diminution des probabilités de rencontre. Ainsi, lorsqu’un peptide est cyclisé en solution, une concentration entre 0,1 et 1 mM est recommandée s’il n’est pas déjà dans une conformation facilitant suffisamment sa condensation intramoléculaire rapide.27, 56 Une autre solution intéressante à ce
problème est la cyclisation du peptide sur support solide. Lorsqu’ils sont ancrés, une séparation physique entre les chaînes peptidiques crée un milieu de pseudo-dilution, offrant un résultat similaire avec beaucoup moins de solvant.57 Plus précisément, une bille de résine fonctionnalisée en surface à un taux de substitution plutôt faible
permet de créer un espace physique constant entre les molécules.58
La synthèse peptidique sur support solide (SPPS) apporte plusieurs avantages par rapport à la synthèse en solution, dont la possibilité d’utiliser plusieurs équivalents des réactifs pour une vitesse de réaction augmentée et une purification simple et rapide par filtration à chaque étape de synthèse.59 Cependant, elle est moins
adaptée pour produire de grosses quantités de produit et les techniques analytiques effectuables sur les molécules ancrées sont limitées. Pourtant, elle reste la méthode la plus courante pour la construction des peptides et est très utilisée pour les cycliser.26, 60, 61
4 La synthèse peptidique sur support solide et les méthodes
d’ancrage associées
Les résines les plus souvent utilisées pour la synthèse peptidique sont à base de polymères de polystyrène réticulé, ce qui offre un gonflement de la résine suffisant dans les solvants polaires aprotiques utilisés lors des couplages peptidiques, comme le DCM et le DMF, à un faible coût. Le gonflement d’une résine est le paramètre le plus important à considérer pour son utilisation, puisqu’il influence directement l’exposition des fonctions à sa surface au solvant et donc leur réactivité.62, 63 Cependant, l’hydrophobicité de ces résines ne convient pas à
toutes les synthèses et l’utilisation de résines plus polaires à base de polyéthylène glycol (PEG), comme la ChemMatrix®, permet la préparation de peptides plus complexes64 ainsi que la possibilité de travailler avec des
solvants plus polaires comme le méthanol ou l’eau.65 En revanche, ces résines sont beaucoup plus
dispendieuses que celles à base de polystyrène.
Un autre paramètre important d’un support solide est son taux de substitution, ce qui représente la proportion de fonctions modifiables à la surface des billes de résine par rapport à leur masse. Un taux de substitution élevé (≥0,8 mmol/g) permet d’obtenir plus de produit par quantité de résine, mais le degré d’interactions entre les sites d’ancrage sera plus élevé que pour une résine à taux de substitution faible (≤0,4mmol/g), donc la pseudo-dilution sera moins efficace. Plusieurs méthodes de mesure du taux de substitution d’une résine existent selon la fonction présente à sa surface. La plus commune lors de synthèse peptidique sur support solide est le dosage du groupement protecteur Fmoc.66 Son clivage par une solution basique, généralement de la pipéridine ou du
DBU, génère du dibenzofulvène qui peut être dosé par absorbance UV.67 L’absorbance obtenue, la masse de
la résine clivée, le volume de la solution dosée et le coefficient d’absorption molaire à la longueur d’onde observée, permettent de calculer le taux de substitution à l’aide d’une équation (Eq 1) dérivée de la loi de Beer-Lambert (Eq 2).68, 69 Le DBU est généralement préféré à la pipéridine pour le dosage, puisqu’il ne forme pas
d’adduit avec le dibenzofulvène qui peut faire varier l’absorbance mesurée.67 Le dosage d’une résine permet
non seulement de vérifier le degré de pseudo-dilution, mais aussi de connaître la quantité de peptide sur la résine et le rendement d’un couplage. Une faible quantité de résine, environs 25 mg est requise pour le dosage.
(eq 1)
𝑆𝑆 = 𝐴𝐴𝑉𝑉𝑚𝑚1𝑉𝑉𝑘𝑘 2
(eq 2) 𝐴𝐴 = 𝜀𝜀𝜀𝜀𝜀𝜀
Où S = Taux de substitution (mmol/g) A = Absorbance
V1 = Volume de la première dilution (ml) m = Masse de résine (mg)
k = Constante relative au coefficient d’absorption molaire, à la longueur d’onde observée et aux dilutions effectuées (mmol)
V2 = Volume transféré dans la seconde dilution (ml) ε = Coefficient d’absorption molaire (M-1cm-1) c = Concentration (M)
Un autre aspect crucial de la SPPS, encore plus dans l’optique d’une macrocyclisation subséquente, est le choix des groupements protecteurs et leur orthogonalité. Sans groupements protecteurs, la synthèse peptidique serait impossible, puisque les couplages seraient imprévisibles. Par exemple, un acide aminé libre mis dans des conditions de couplage normal formerait de nombreuses tailles de polymères sans aucun contrôle. Pour remédier à ce problème, toutes les fonctions nucléophiles et électrophiles, sauf l’acide et l’amine à coupler, sont protégées pour masquer leur réactivité et pour permettre de les révéler à nouveau lorsque désiré. Le concept d’orthogonalité réfère à la possibilité de cliver un groupement protecteur sélectivement par rapport à un autre, permettant ainsi de ne révéler que les fonctions voulues. Pour effectuer une synthèse peptidique classique sur support solide, deux degrés d’orthogonalité sont nécessaires.27 Deux grandes familles existent pour répondre à
ce besoin, soient la chimie Fmoc/tbu et Boc/Bn, pour une déprotection sélective en milieu basique/acide et acide/HF respectivement.70 Puisque la chimie Fmoc/tbu est désormais la plus commune par sa simplicité, son
efficacité, la disponibilité des réactifs et leur coût raisonnable, elle sera la seule méthode discutée.
R1 OH O N N N O N N PF6 Base N N N O R1 O O N N R1 O O N N N N N O R2 NH2 R1 NH O R2 HATU N N N HO
Figure 10 Mécanisme d’activation d’acide et de couplage peptidique avec une amine pour l’agent de couplage HATU.
Chaque acide aminé à ajouter à la chaîne est protégé par un Fmoc sur l’amine (Nα) et par un groupement labile en milieu acide sur la chaîne latérale pouvant varier selon la fonction. La fonction acide libre est alors couplée au support solide, généralement à une amine ou un alcool, à l’aide d’un agent de couplage et d’une amine tertiaire comme base encombrée dans le DMF ou le DCM. Une gamme importante d’agents de couplage est commercialement disponible et le mécanisme de couplage du HATU, l’un des plus performants pour la formation de liens amide, est décrit dans la Figure 10.40-42 Une fois le couplage complété, le groupement Fmoc peut être
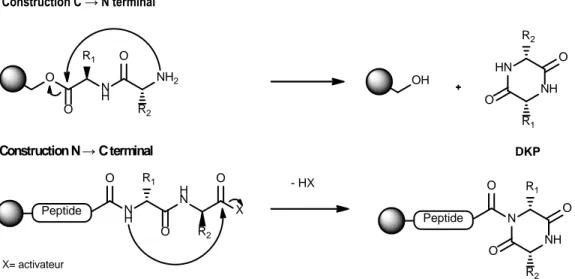
clivé par une solution de pipéridine, puis le prochain couplage peut être effectué. Une fois le peptide complet, une déprotection des chaînes latérales et un clivage du peptide de la résine simultanés à l’aide d’une solution de TFA permettent d’obtenir le peptide libre.69, 71 Le choix synthétique de construction du peptide du C- au
N-terminal vient des nombreuses difficultés rencontrées, comme une racémisation importante, lors de couplages inverses avec l’acide sur le support solide.72 De plus, la formation de dicétopipérazine (DKP) est plus fréquente
H N X O R2 O N H R1 N H O NH2 R2 O R1 O O Peptide OH HN NH O O R2 R1 DKP N NH O O R1 R2 O Peptide X= activateur - HX
Figure 11 Mécanisme de formation de DKP lors d’une synthèse peptidique sur support solide en sens classique et en sens inverse.
La formation de DKP est aussi une considération importante dans le sens classique de construction, surtout lorsqu’il est question du choix d’ancrage au support solide. En effet, si la fonction d’ancrage forme un ester peu encombré, le risque de formation de DKP et donc de clivage prématuré augmente lors du second couplage, surtout si l’un ou l’autre des acides aminés est peu encombré ou induit une conformation cis du lien amide intermédiaire.74, 75
L’ancrage d’un peptide est d’ailleurs un paramètre très important à considérer, surtout en prévision d’une macrocyclisation. En effet, alors que pour une construction linéaire, le peptide est généralement ancré par son acide terminal, cette option n’est pas toujours possible pour un macrocycle, surtout lorsque le cycle à former implique cet acide. Ainsi, un ancrage par une chaîne latérale ou par un amide du squelette est normalement envisagé. Pour un ancrage par une chaîne latérale, l’une d’elle doit contenir une fonction acide,76, 77 amine,78
alcool,79 guanidine80 ou indole81 et pourra être couplée à une ancre correspondante sur la résine pour créer une
fonction labile, généralement dans les mêmes conditions acides que celles utilisées pour cliver un peptide linéaire.
Quant à l’ancrage par l’amide, la méthodologie BAL est la plus courante. Une amination réductive est effectuée entre l’amine du premier acide aminé et un tri-alkoxybenzaldéhyde sur support solide, formant une amine benzylique secondaire. Par couplage entre cette amine et le second acide aminé, la chaîne peut être continuée (Figure 12).82 Puisque l’amide formée est tertiaire, elle induit une orientation cis ce qui aide à la macrocyclisation
future, mais augmente les risques de formation de DKP.83 Un clivage en milieu acide est alors possible pour
libérer l’amide secondaire une fois le peptide complété.84 Dans tous les cas, un troisième degré d’orthogonalité
est nécessaire pour effectuer une macrocyclisation sur support solide. Pour ce faire, les groupements Allyl/Alloc, comme protecteurs d’acides et d’amines, respectivement, sont de bons candidats et ont été amplement
Construction C → N terminal
étudiés.27, 85, 86 Leur clivage par un complexe de palladium et un nucléophile est presque quantitatif et est
orthogonal aux autres groupements protecteurs et aux méthodes d’ancrages mentionnées.
O O H OMe OMe O N H OMe OMe R1 O O PG O N OMe R1 O O PG O HN R2 Fmoc OMe H2N R1 O O PG NaBH3CN HO O R2 H N Fmoc HATU, NMM Accroche BAL
Figure 12 Installation de deux acides aminés sur support solide à l’aide de la méthode d’ancrage BAL.
Une dernière méthode d’ancrage, analogue à la stratégie BAL, permet de créer en une seule étape un amide tertiaire lié à un trialkoxybenzaldéhyde sur résine via une réaction multicomposante de Ugi (Figure 13). Les mêmes conditions de clivage et les mêmes groupements protecteurs peuvent être utilisés pour cette méthode, ce qui la rend plus rapide pour la même efficacité.87 De plus, une flexibilité dans le choix des méthodes
d’ancrages possibles permet de synthétiser plus de peptides cycliques complexes en s’adaptant ainsi à leurs difficultés. O O H OMe OMe O N OMe R1 O O PG O HN R2 Fmoc OMe H2N R1 O O PG HO O R2 H N Fmoc Accroche BAL tBuNC O NH tBu
Figure 13 Installation de deux acides aminés sur support solide à l’aide la méthode d’ancrage Ugi analogue à la méthode BAL. Voir la figure 14 pour le mécanisme de formation du dipeptide.
5 Les réactions multicomposantes
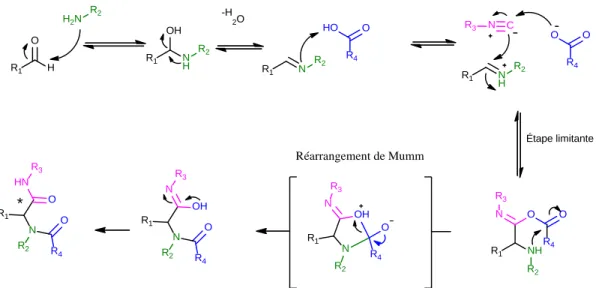
La réaction de Ugi est une réaction multicomposante (MCR) nécessitant quatre synthons, soit une amine, un acide carboxylique, un aldéhyde et un isonitrile. Leur combinaison donne un motif peptidomimétique, donc la formation de deux liens amides séparés d’un carbone α-substitué. Le mécanisme de la réaction est illustré dans la Figure 14, où l’on peut voir le réarrangement de Mumm final.88 Cette réaction a été explorée dans le domaine
de la synthèse peptidique. À titre d’exemple, pour lier deux fragments peptidiques ensemble de manière similaire à une synthèse par fragments de peptides complexes,89 pour une macrocyclisation,90 pour la formation complète
de macrocycles peptidiques91, 92 ou, comme mentionné plus tôt, pour attacherr un peptide tout en effectuant le
premier couplage.87 Puisque la substitution tertiaire d’un des amides créés est un cis-orienteur, cette réaction a
R2 H2N R1 H O R1 N H OH R2 R1 N R2 HO R1 N H R2 O C N R3 R1 NH N R3 O R2 O R1 N N R3 OH R2 O R1 N N R3 OH R2 O R1 * N HN R3 O R2 O -H 2O O R4 R 4 O R4 R4 R4 R4 Réarrangement de Mumm Étape limitante
Figure 14 Mécanisme de la réaction de Ugi. Le carbone chiral formé est identifié par une étoile.
Un autre avantage plus général aux réactions multicomposantes qui les rend si populaires en ce moment dans tous les domaines est leur très grande puissance exploratoire. En effet, le nombre de produits formés par les diverses combinaisons de plusieurs synthons est exponentiel. Ainsi, avec par exemple quatre options pour chaque synthons, le nombre de possibilités totale est de 64 molécules différentes. Cette caractéristique a, par exemple, été utilisée dernièrement avec la réaction de Ugi pour créer une chimiothèque exploratoire de benzodiazépines et donc pour découvrir de nouveaux composés bioactifs à optimiser.93
De plus, la création de plusieurs liens dont un entre deux carbones en une seule étape ainsi que l’économie d’atomes importante vu la libération d’une molécule d’eau seulement rendent la réaction de Ugi très attrayante en chimie pharmaceutique et en chimie verte.94 Cependant, le plus gros obstacle de cette réaction est la création
d’un centre chiral en proportions racémiques, ce qui limite son potentiel, c’est pourquoi de nombreux travaux sont aujourd’hui orientés vers des variations pour tenter d’obtenir un produit énantiopur. Cet exploit a d’ailleurs été complété dernièrement grâce à un catalyseur d’acide phosphorique chiral.95 En effet, la stéréochimie est
souvent cruciale pour la reconnaissance des peptides par leur cible protéique, donc un mélange racémique rend la moitié du produit inadéquat dans ces circonstances.
5.1 La réaction de Ugi-Smiles
Le grand potentiel de la réaction de Ugi a poussé certains groupes à diversifier davantage la réaction en substituant l’une des quatre fonctions requises, le plus souvent l’acide, par une fonction de réactivité similaire pour permettre encore davantage de flexibilité. Ugi lui-même en a exploré plusieurs, dont l’utilisation d’azoture d’hydrogène (HN3),96 d’eau (H2O), d’acide thiosulfurique (H2S2O3), et de séléniure d’hydrogène (H2Se),97 tandis
substitution est complexe, puisque l’acide participe de différentes manières à presque toutes les étapes du mécanisme, donc le substitut doit avoir une réactivité très similaire.100
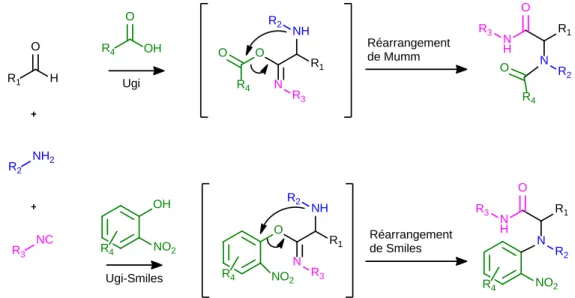
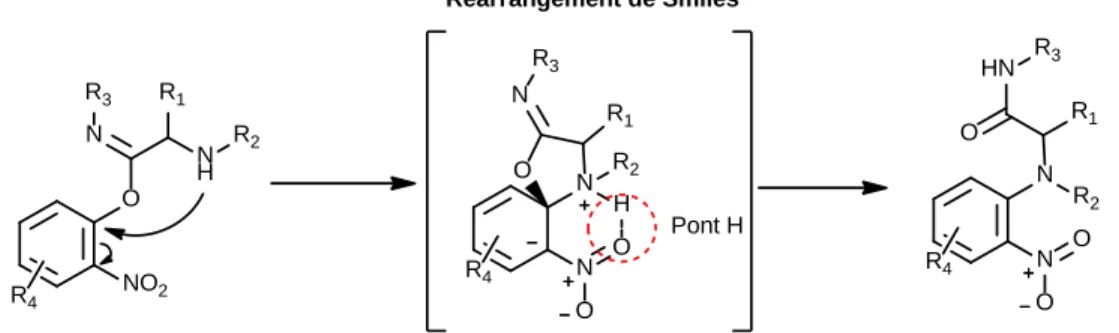
La substitution avec le nitrophénol, appelée Ugi-Smiles pour son réarrangement de type Smiles au lieu de Mumm (Figure 15),100, 101 est intéressante puisqu’elle permet de former des anilines, un motif assez recherché
en chimie médicinale.102 Du côté du mécanisme, la réaction est presque identique à la réaction de Ugi, si ce
n’est du réarrangement final. D’ailleurs, le réarrangement de Smiles a une énergie d’activation beaucoup plus importante que celui de Mumm, si bien qu’il constitue une deuxième barrière énergétique limitant la vitesse de réaction, contrairement à la réaction de Ugi où seule l’attaque nucléophile de l’isonitrile sur l’imine est limitante.103
En effet, l’énergie d’activation de l’attaque nucléophile est de 19,8 et 15,8 kcal/mol pour la Ugi et la Ugi-Smiles, respectivement, contre 1,0 versus 11,7 kcal/mol pour le réarrangement.100 Cette différence énergétique se
traduit, entre autres, par une nécessité de chauffer les réactions de Ugi-Smiles pour qu’elles fonctionnent, généralement à au moins 40 ou à 60°C.104 L’utilisation des micro-ondes pour accélérer la réaction a aussi été
tentée et s’est révélée fort efficace dans certaines situations.105
R1 H O R2 NH2 R3 NC R4 OH O OH NO2 R4 O N R3 O R4 R1 NH R2 O N R3 R1 NH R2 NO2 R4 N H O R3 R1 N R2 O R4 N H O R3 R1 N R2 NO2 R4 Ugi Ugi-Smiles Réarrangement de Smiles Réarrangement de Mumm
Figure 15 Réactions de Ugi et de Ugi-Smiles selon leurs principales différences mécanistiques.
Beaucoup de travail a été effectué pour déterminer les possibilités et les limitations de la réaction de Ugi-Smiles. Les variantes intéressantes seront discutées pour chaque synthon. Premièrement, les aldéhydes aliphatiques et benzyliques, tout comme les cétones aliphatiques, peuvent être utilisées. Les aldéhydes aliphatiques sont les plus réactifs, apportant une énergie d’activation plus faible que les aldéhydes benzyliques, probablement dû à la perte de conjugaison entre le carbonyle et le cycle aromatique lors de l’attaque nucléophile de l’isonitrile. Ainsi, une température plus importante est nécessaire avec les aldéhydes benzyliques pour obtenir des rendements similaires. Pour ce qui est des cétones, leur électrophilicité plus faible les rendent moins réactives,