Structure et dynamique du peptide de fusion
membranaire du virus Influenza et son impact
sur la membrane
Thèse Sébastien Légaré Doctorat en biochimie Philosophiæ doctor (Ph.D.) Québec, Canada © Sébastien Légaré, 2014Résumé
La fusion membranaire est une étape essentielle du cycle infectieux du virus Influenza dont la compréhension est actuellement incomplète. La fusion nécessite la protéine de surface virale hémagglutinine et, en particulier, ses vingt acides aminés N-terminaux formant le peptide de fusion. Ce peptide a été démontré capable d’initier la fusion membranaire même lorsque séparé du reste de la protéine, mais le mécanisme moléculaire par lequel il y parvient reste méconnu. Afin de mieux comprendre ce mécanisme, nous avons effectué des simulations de dynamique moléculaire du peptide de fusion, du mutant fusogène F9A et du mutant non fusogène W14A, dans des membranes modèles.
Dans un premier temps, nous avons étudié la structure et la dynamique du peptide de fusion. Le peptide de fusion a adopté des conformations en hélice-α complète et en coude, et s’est positionné à l’interface membranaire presque parallèlement à la surface de la membrane. Les peptides mutants ont en plus adopté une structure en épingle. La dynamique des peptides a donc été associée à celle d’un V flexible, changeant de conformation par des mouvements de charnière. Dans un second temps, les perturbations membranaires induites par les peptides ont été étudiées par simulations. Ces perturbations incluent la protrusion des chaînes lipidiques et l’intrusion des têtes polaires. Ces deux perturbations ont été causées par des ponts hydrogène entre les phosphates lipidiques et les amides N-terminales des peptides s’insérant sous la surface de la membrane. La quantité d’intrusion des têtes polaires induite par les mutants en simulation était corrélée à leur activité fusogène expérimentale et à la profondeur d’insertion de leur extrémité N-terminale.
Suivant ces résultats, nous proposons que l’intrusion des têtes polaires complémente la protru-sion des chaînes lipidiques lors de la fuprotru-sion membranaire en réduisant les forces répulsives entre les têtes polaires des membranes juxtaposées. Ce mécanisme modèle de fusion membranaire pourra avoir un impact sur les futures recherches d’antiviraux contre Influenza.
Abstract
Membrane fusion is an essential step of the Influenza virus infectious cycle whose understand-ing remains incomplete. Fusion requires surface viral protein hemagglutinin and, in particular, its twenty N-terminal amino acids composing the fusion peptide. This peptide was shown to initiate fusion even when isolated from the rest of the protein, but the molecular mechanism by which it achieves membrane fusion is still misunderstood. To better understand this mech-anism, we performed molecular dynamics simulations of the fusion peptide, fusogenic F9A mutant and nonfusogenic W14A mutant, in model membranes.
First, we studied the structure and dynamics of the fusion peptide. The fusion peptide adopted straight α-helical and kinked conformations, and inserted at the membrane interface with an almost parallel orientation with the membrane surface. Mutant peptides additionaly adopted a hairpin structure. The dynamics of the peptides was hence compared to that of a flexible V, switching conformation by hinge movements. In a second step, fusion peptide-induced mem-brane perturbations were studied from simulations. Those perturbations include lipid tail protrusion and polar head intrusion. The two perturbations were caused by hydrogen bonding between lipid phosphates and membrane inserted peptide N-terminal amides. The amount of polar head intrusion induced by the mutant peptides in simulations was correlated to their experimental fusogenic activity and the insertion depth of their N-termini.
Following those results, we propose that polar head intrusion would complement lipid tail protrusion during membrane fusion by reducing the repulsive forces between juxtaposed mem-branes polar heads. This model of membrane fusion mechanism may have an impact on future Influenza antiviral research.
Table des matières
Résumé iii
Abstract v
Table des matières vii
Liste des tableaux ix
Liste des figures xi
Liste des abréviations xiii
Remerciements xix Avant-propos xxi 1 Introduction 1 Introduction 1 1.1 L’Influenza . . . 1 1.2 Le virus Influenza. . . 5 1.3 Contrer l’Influenza . . . 12
1.4 Pharmaco-économie et justification du projet . . . 16
1.5 La fusion membranaire . . . 19
1.6 L’hémagglutinine . . . 29
1.7 Le peptide de fusion membranaire . . . 35
2 Objectifs 41 2.1 La dynamique du peptide de fusion dans les membranes . . . 41
2.2 Les perturbations lipidiques induites par le peptide de fusion . . . 41
3 Méthode 43 3.1 Dynamique moléculaire . . . 43
3.2 Matériel et logiciels . . . 44
3.3 Préparation des simulations . . . 45
3.4 Théorie de la DM. . . 47
3.5 Paramètres de simulation . . . 56
4 The fusion peptide flexible flat V conformation 67
4.1 Résumé . . . 67
4.2 Abstract . . . 68
4.3 Introduction. . . 68
4.4 Methods . . . 69
4.5 Results and Discussion . . . 71
4.6 Conclusion . . . 79
4.7 Acknowledgements . . . 80
4.8 Supporting Material . . . 80
4.9 Annexe . . . 95
5 Perturbations lipidiques 97 5.1 Distribution des lipides autour de PF. . . 97
5.2 Adaptation des chaînes lipidiques à la présence du peptide . . . 98
5.3 Ordre des lipides . . . 99
5.4 Amincissement de la membrane . . . 101
5.5 Lien avec la fusion membranaire . . . 102
6 The fusion peptide promotes lipid polar head intrusion 103 6.1 Résumé . . . 104
6.2 Abstract . . . 104
6.3 Introduction. . . 105
6.4 Methods . . . 106
6.5 Results and Discussion . . . 107
6.6 Conclusion . . . 117
6.7 Acknowledgements . . . 118
6.8 Supporting Information . . . 119
7 Discussion 129 7.1 Comportement du peptide de fusion en simulations de DM. . . 129
7.2 Comparaisons avec les données expérimentales. . . 132
7.3 Comparaison avec les travaux de DM précédents . . . 134
7.4 Implications des travaux sur la dynamique du peptide . . . 135
7.5 Perturbations membranaires . . . 136
7.6 Protrusion des chaînes lipidiques . . . 136
7.7 Intrusion des têtes polaires. . . 139
7.8 Impacts du projet . . . 142
7.9 Perspectives . . . 143
8 Conclusion 147 A Fichier de configuration NAMD 149 A.1 Description des paramètres de simulation . . . 151
B Scripts Tcl pour analyses dans VMD 155
Liste des tableaux
1.1 Rentabilité des interventions contre Influenza . . . 18
4.1 Total, partial and individual SASA . . . 84
4.2 Occurrence of backbone hydrogen bonds in the kink region of the fusion peptide . 87
6.1 Protrusion and intrusion statistics at the N-terminus of the three peptides . . . 110
6.2 Amino acid sequence of the WT FP and the two studied mutant peptides . . . 119
7.1 Probabilité d’intrusion pour les lipides liés au N-terminus des trois peptides en différentes conformations. . . 141
Liste des figures
1.1 Distribution géographique des infections Influenza . . . 2
1.2 Microphotographie de virus Influenza de type A . . . 6
1.3 Schéma du virus Influenza A . . . 7
1.4 Cycle de réplication du virus Influenza . . . 8
1.5 Nombre d’infections lors de la pandémie de 2009 et nombre projeté sans vaccination 13 1.6 Structure de l’arbidol et interaction proposée avec les phospholipides membranaires 16 1.7 Structure de phospholipides d’intérêt . . . 20
1.8 Phases lipidiques . . . 21
1.9 Phases lamellaires gel Lβ et cristal liquide Lα . . . 22
1.10 Membrane cellulaire eucaryote . . . 23
1.11 Modèles membranaires . . . 24
1.12 Densité des différentes régions des bicouches lipidiques . . . 26
1.13 Profil de pression latérale des bicouches . . . 27
1.14 Fusion membranaire . . . 28
1.15 Structure trimérique et sous-unités de l’hémagglutinine. . . 29
1.16 Structure d’un monomère d’hémagglutinine . . . 31
1.17 Acide N-acétylneuraminique . . . 32
1.18 Changement conformationnel pH-dépendant de l’hémagglutinine . . . 34
1.19 Participation de l’hémagglutinine à la fusion membranaire . . . 35
1.20 Structures du PF en « V » inversé . . . 36
1.21 Structures alternatives du PF . . . 37
1.22 Fusion membranaire mesurée par le mélange de lipides à pH acide et neutre . . . . 38
1.23 Structure du PF à pH 7,4 et 5 . . . 39
1.24 Effet du peptide sur les membranes phospholipidiques . . . 39
3.1 Schéma de la construction du système simulé . . . 46
3.2 Équivalences entre les interactions fondamentales et les termes de champ de force . 49 3.3 Illustration des termes d’interaction inclus dans le champ de force CHARMM27r . 51 3.4 Énergie d’interaction de type Lennard-Jones entre deux atomes de carbone . . . . 57
3.5 Schéma des frontières périodiques . . . 59
4.1 Side views of the straight helix and kinked conformations of the WT fusion peptide 72 4.2 Side chain insertion depths under the phosphates per residue . . . 73
4.3 Top-view 20 conformations of the wild-type, F9A, and W14A fusion peptides . . . 75
4.4 Backbone hydrogen bond patterns . . . 81
4.5 Average1Hα chemical shift differences for the WT peptide . . . 82
4.7 Side chains insertion depths under the phosphate groups from WT simulations . . 85
4.8 Average helical fraction for the three peptides and pdb 1IBN . . . 86
4.9 Average1Hα chemical shifts differences for the F9A and W14A mutants . . . 88
4.10 Distances between α protons and aromatic rings in the region of the FP kink . . . 89
4.11 Standard deviation σ of the backbone Φ dihedral angle per residue . . . 90
4.12 WT kink angle time series . . . 91
4.13 F9A mutant kink angle time series . . . 92
4.14 W14A mutant kink angle time series . . . 93
4.15 Kink angle θ distributions for the three peptides . . . 94
5.1 Distribution des lipides autour du PF . . . 98
5.2 Vue en coupe du réarrangement structural des chaînes lipidiques autour du PF . . 99
5.3 Paramètres d’ordre SCD expérimentaux et par DM . . . 100
5.4 Changements de SCD des chaînes palmitoyl et oleoyl en contact avec le PF par DM101 5.5 Épaisseur de la membrane . . . 102
6.1 Normalized probability of aliphatic carbon protrusion surrounding the peptides . . 108
6.2 Typical H-bonding pattern between a phosphate and the peptide. . . 109
6.3 Normalized probability of polar head intrusion surrounding the peptides . . . 111
6.4 Bilayer snapshots highlighting polar head intrusion and lipid tail protrusion . . . . 113
6.5 Membrane invagination at the FP N-terminus . . . 114
6.6 Depths of insertion of the peptide backbone amides under the phosphates . . . 115
6.7 Proposed model for FP-catalyzed lipid mixing based on lipid head intrusion . . . . 117
6.8 Peptide RMSDs from all WT simulations . . . 120
6.9 Peptide RMSDs from all F9A simulations . . . 121
6.10 Peptide RMSDs from all W14A simulations . . . 122
6.11 Frequencies and average times of protrusion events for the WT FP . . . 123
6.12 Frequencies and average times of protrusion events for the F9A mutant. . . 124
6.13 Frequencies and average times of protrusion events for the W14A mutant . . . 125
6.14 Normalized probability of aliphatic carbon protrusion in the opposite leaflet . . . . 126
6.15 Normalized probability of polar head intrusion in the opposite leaflet . . . 126
6.16 Average position of lipid carbons and phosphates . . . 127
Liste des abréviations
BMRB BioMagResBank CD Dichroïsme circulaire
CHARMM Chemistry at Harvard macromolecular mechanics CMAP Carte des termes croisés Φ/Ψ
DEER Résonance électron-électron double DM Dynamique moléculaire
DHPC Dihexanoyl-phosphatidylcholine DOPC Dioleoyl-phosphatidylcholine DPC Dodécyl-phosphatidylcholine DPPS Dipalmitoyl-phoshatidylsérine FDA Food and drug administration
FTIR Spectroscopie infrarouge à transformée de Fourier FP Fusion peptide
GSK GlaxoSmithKline HA Hémagglutinine
HAT Protéase de voies respiratoires humaine semblable aux trypsines ICTV Comité international sur la taxonomie des virus
MM Mécanique moléculaire NA Neuraminidase
NAMD Nanoscale molecular dynamics OMS Organisation mondiale de la santé
PACE Protein in atomistic details coupled with coarse-grained environment PC Phosphatidylcholine
PDB Protein data bank
PE Phosphatidyléthanolamine PF Peptide de fusion
PG Phosphatidylglycérol PI Phosphatidylinositol PIB Produit intérieur brut
POPC Palmitoyl-oléoyl-phosphatidylcholine POPE Palmitoyl-oléoyl-phosphatidyléthanolamine POPG Palmitoyl-oléoyl-phosphatidylglycérol POPI Palmitoyl-oléoyl-phosphatidylinositol
PRE Augmentation de relaxation paramagnétique PS Phosphatidylsérine
PSF Protein structure file PSM Palmitoyl-sphingomyéline PTFE Polytétrafluoroéthylène
REDOR Double résonance avec écho rotationnel RMN Résonance magnétique nucléaire
RMSD Écart quatratique moyen, « Root mean square deviation » RNP Ribonucléoprotéique
RPE Résonance paramagnétique électronique SM Sphingomyéline
TM Domaine transmembranaire
TMPRSS2 Protéase à sérine S1 transmembranaire de type 2 VMD Visual Molecular Dynamics
À mon père, le meilleur exemple de vertu que je connaisse
[Pour] une intelligence qui [...] connaîtrait toutes les forces dont la nature est animée, [...] rien ne serait incertain.
Marquis Pierre-Simon Laplace Essai philosophique sur les probabilités, 5eéd. Bachelier, Paris, 1825
Remerciements
Le premier remerciement de cette thèse revient à mon directeur de recherche Patrick Lagüe. Il m’a soutenu tout au long du doctorat et m’a enseigné l’importance de la rigueur scientifique, autant dans l’analyse des simulations de dynamique moléculaire que dans la rédaction et la présentation des résultats. J’espère maintenant pouvoir appliquer cette rigueur au cours de ma future carrière scientifique, et ce, à la hauteur de la formation reçue. Je remercie aussi Patrick de m’avoir procuré le milieu de travail exceptionnel qu’était le laboratoire de son groupe de recherche au sein de l’institut de biologie intégrative et des systèmes (IBIS) de l’université Laval.
Je remercie ensuite les membres de mon comité évaluateur, Michèle Auger, Lisa Topolnik et Louis Dubé, qui m’ont fourni de précieux conseils lors des rencontres de comité. Je suis aussi très reconnaissant de la participation de Michèle et Lisa à mon jury de thèse.
L’évaluation de ma thèse n’aurait pas été possible sans la participation très appréciée de Pierre Lavigne de l’université de Sherbrooke, qui a généreusement accepté d’être le membre externe de mon jury de thèse.
J’exprime également ma gratitude envers le professeur de chimie théorique Thanh-Tung Nguyen-Dang pour avoir répondu à mes questions triviales sur la mécanique quantique et pour avoir supervisé mon projet spécial de photochimie.
Il est important pour moi de remercier le professeur de biophysique François Boucher de l’université du Québec à Trois-Rivières qui m’a permis d’entreprendre un projet de dynamique moléculaire à la fin du baccalauréat et de confirmer mon intérêt pour ce domaine d’études. Je tiens à remercier mes collègues de laboratoire avec qui j’ai passé d’innombrables bons moments, Xavier Barbeau, Barbara Collignon, Richard Daigle, Olivier Fisette, Luc Grenier, Jean-Francois Rheault, Julie-Anne Rousseau et Andrei Tudor. Je remercie aussi les stagiaires qui ont participé au dynamisme du laboratoire, Christelle Bidouille, Joël Tremblay-Marchand, Alexandre Marquis et Jean-Sébastien Milanese.
J’envoie un gros merci à ma famille. La sagesse de mon père et l’intelligence de ma mère m’ont inspiré la soif de vérité qui m’a poussé à poursuivre des études de cycle supérieur en
sciences fondamentales. Mon frère, le philosophe, avec qui j’espère un jour pouvoir mettre en application certains des nombreux projets tirés de nos discussions.
Merci à ma chérie Francesca, qui comprend mieux que quiconque mes succès et mes échecs. Sa présence a allégé la complétion de ma thèse et continue d’égayer mes journées.
Finalement, il est important de souligner le support financier obtenu et sans lequel cette thèse n’aurait pu être complétée. Les organismes à remercier pour cette thèse sont le FRQNT, PROTEO, Calcul Canada, le CRSNG, la FCI et le département de biochimie, microbiologie et bio-informatique de l’université Laval.
Avant-propos
Les principaux résultats présentés dans cette thèse ont été publiés sous forme d’articles scien-tifiques dans des journaux avec comité de lecture. Les deux publications rédigées, dont je suis le premier auteur, sont insérées aux chapitres 4et6de la thèse avec autorisation des éditeurs des journaux respectifs.
Chapitre 4 : Structure et dynamique du peptide de fusion
L’article constituant ce chapitre présente une analyse de la dynamique du peptide de fusion en simulations de dynamique moléculaire en comparaison à la dynamique suggérée par différentes méthodes expérimentales. Cet article est publié sous la référence :
Sébastien Légaré et Patrick Lagüe. The Influenza Fusion Peptide Adopts a Flexible Flat V Conformation in Membranes. Biophys. J., 102 :2270–2278, 2012,
doi : 10.1016/j.bpj.2012.04.003
Le Pr. Patrick Lagüe et moi-même avons entièrement rédigé cet article. J’ai effectué toutes les simulations et analyses qui y sont présentées, sous la supervision du Pr. Lagüe. L’article est inclu dans la thèse avec l’autorisation d’Elsevier.
Chapitre 6 : Intrusion des têtes polaires
L’article constituant ce chapitre présente l’intrusion des têtes polaires, une perturbation lipi-dique induite par le peptide de fusion et décrite pour la première fois dans ces travaux. Cet article est publié sous la référence :
Sébastien Légaré et Patrick Lagüe. The influenza fusion peptide promotes lipid polar head intrusion through hydrogen bonding with phosphates and N-terminal membrane insertion depth. Proteins, 82 :2118-2127, 2014, doi : 10.1002/prot.24568
Le Pr. Patrick Lagüe et moi-même avons entièrement rédigé cet article. J’ai effectué toutes les simulations et analyses qui y sont présentées, sous la supervision du Pr. Lagüe. L’article est inclu dans la thèse avec l’autorisation de John Wiley and Sons.
Chapitre 1
Introduction
1.1
L’Influenza
L’Influenza, aussi nommée grippe, est une infection virale dont les symptômes ont déjà été ressentis par la vaste majorité des individus de la planète. Ces symptômes se rapprochent de ceux du rhume : fièvre, toux, congestion nasale, maux de gorge et de tête, fatigue et myal-gie [1]. Une infection à l’Influenza est habituellement éliminée naturellement par le système immunitaire et la durée des symptômes dépasse rarement une semaine [2,3]. Cependant, une faible proportion des infections mène à une hospitalisation ou même au décès de l’individu atteint. De plus, un taux de mortalité particulièrement élevé a été enregistré lors de certaines pandémies du dernier siècle [4].
Tel qu’illustré à la figure 1.1, l’Influenza se retrouve pratiquement partout autour du globe. Le taux d’infection varie cependant selon les régions puisque la transmissibilité du virus est diminuée par les hauts taux d’humidité ou les températures dépassant 30◦C [5]. Dans les régions tempérées, les infections sont plus fréquentes durant la saison hivernale, soit au mois de janvier dans l’hémisphère nord et au mois de juillet dans l’hémisphère sud. À l’équateur, le nombre d’infections est plus également distribué au cours de l’année [6].
1.1.1 La grippe saisonnière
Il est estimé qu’environ 10% de la population mondiale est affectée par l’Influenza saisonnière chaque année, et 10% à 20% dans les pays développés [7]. Chaque individu nord-américain contracterait donc la grippe en moyenne une fois tous les 5 à 10 ans. Les taux d’infection sont particulièrement élevés chez les enfants d’âge scolaire, ce qui contribue de manière importante à la transmission d’Influenza dans les foyers [8]. Les infections ne sont fatales que dans de faibles proportions (∼ 0,05%) [7]. Les décès ne sont habituellement pas directement causés par l’Influenza, mais plutôt par des complications dues à l’affaiblissement de l’individu par le virus Influenza. Le nombre total de décès reliés à l’Influenza est donc estimé entre 300 000 et
Figure 1.1: Distribution géographique des infections Influenza. La taille des points rouges est proportionnelle au taux d’infectivité. Le nombre de points dépend du nombre de centres de détection et n’est donc pas indicateur du nombre d’infections. Adapté de [6].
500 000 par année [7, 9], soit environ 0,005% de la population mondiale. En comparaison, le tabac cause 10 fois plus de décès, environ 6 millions dans le monde. Au Canada, on estime le nombre de décès reliés à l’Influenza entre 2000 et 8000 par an, soit ∼ 0,015% de la population, un pourcentage très près de celui estimé aux États-Unis. 90% des décès reliés à l’Influenza saisonnière surviennent chez des individus de 65 ans et plus [10].
Il est difficile de mesurer l’entièreté de l’impact de l’Influenza sur la société humaine. Du point de vue des individus, l’impact d’un épisode de grippe se ressent par quelques jours d’inconfort, parfois important, et de faibles chances de décès. Du point de vue de la société, l’Influenza implique aussi de larges coûts. Le traitement des personnes infectées entraîne des frais directs pour le système hospitalier, mais la majorité des coûts sont indirects et proviennent de la perte de productivité chez les gens atteints [11]. Le fardeau économique total causé par l’Influenza saisonnière aux États-Unis a été évalué à 87 milliards $ annuellement [11]. Considérant que la population du Canada est environ un dixième de celle des États-Unis, on peut estimer que les coûts reliés à l’Influenza saisonnière seraient de ∼ 9 milliards $ par an au Canada, soit presque 1% du produit intérieur brut (PIB). Cette évaluation dépend fortement du nombre d’infections, qui est surestimé selon certains auteurs [12].
1.1.2 Les pandémies d’Influenza
En plus de la grippe saisonnière, des pandémies de grippe surviennent 3 à 4 fois par siècle. Du-rant le dernier siècle, les pandémies de la grippe espagnole, asiatique, de Hong Kong [4,13] et de H1N1 ont eu lieu en 1918, 1957, 1968 et 2009 respectivement. Par définition, une pandémie s’étend sur un plus large territoire qu’une épidémie et touche une grande portion de la popu-lation. La pire pandémie d’Influenza répertoriée est celle de la grippe espagnole de 1918, ayant causé de 20 à 50 millions de décès [7], soit environ 3% de la population mondiale de l’époque. De plus, les décès de cette grippe survenaient particulièrement chez les jeunes adultes. Les recherches pour comprendre quels facteurs ont rendu cette souche particulièrement virulente sont toujours en cours [13]. Les hypothèses actuelles sont que cette souche d’Influenza causait une surstimulation du système immunitaire. Les antibiotiques n’étant pas encore couramment utilisés en 1918, plusieurs décès causés par infections bactériennes secondaires auraient proba-blement aussi été évités si cette pandémie survenait aujourd’hui.
Une pandémie d’Influenza de sérotype H1N1 est survenue en 2009 et a été l’objet d’une vaste couverture médiatique. La signification du sérotype H1N1 sera détaillée à la section
1.2.1. À bien des égards, la pandémie a causé beaucoup moins de dommages qu’attendu avec 18156 décès confirmés dans le monde, dont 428 au Canada [14], et 108 au Québec [15]. En comparaison, les prévisions de l’Organisation Mondiale de la Santé (OMS) datant de 2005 suggéraient que la prochaine pandémie pourrait faire de 1,5 à 3,5 millions de morts dans le monde [7]. Le nombre de décès confirmé est donc environ 100 fois moindre que le nombre attendu. Selon les données recueillies au Québec, cependant, 60% de ces décès ont eu lieu chez des individus âgés de moins de 65 ans [15], en comparaison à 10% pour la grippe saisonnière [10]. Il y a eu 40185 cas d’infection confirmés au Canada [14], ce qui représente ∼ 0,12% sur la population canadienne. Ceci est un taux d’infection bien moindre que celui estimé pour la grippe saisonnière qui est de 10% à 20%. Il est cependant important de faire la distinction entre le nombre de cas confirmés et le nombre estimé. Le nombre de cas estimé tente d’inclure le grand nombre d’infections non répertoriées étant donné la nature souvent bénigne des symptômes de l’Influenza. Au Canada, il est estimé qu’en réalité, la proportion de la population infectée par la souche H1N1 de 2009 aurait été de 16% [15] à 24% [16] selon les études, soit environ 7 millions de personnes. Cela impliquerait cependant que le taux de fatalité de cette souche de H1N1 soit 10 fois moindre de celui des grippes saisonnières. À la lumière des événements de 2009, il a été proposé que le nombre de décès associés à H1N1 soit sous-estimé en omettant des décès de causes secondaires, ou que le nombre estimé de décès causés par les grippes saisonnières soit surestimé [12].
1.1.3 Historique et étiologie
L’humain moderne, soit l’espèce taxonomique homo sapiens, est apparu il y a environ 200 000 ans [17]. Le genre Homo, comprenant entre autres les espèces homo sapiens et homo neander-thalensis, est quant à lui apparu il y a un peu plus de 2 millions d’années. Les populations de chasseurs-cueilleurs étaient alors probablement déjà touchées par des maladies infectieuses semblables à celles connues aujourd’hui. L’Influenza telle qu’on la connaît aujourd’hui néces-site cependant une population humaine dense, et est donc probablement apparue il y a environ 11 000 ans, alors que l’agriculture se développait [18]. La cause de cette maladie était alors certainement ignorée.
L’utilisation du terme Influenza serait d’origine italienne et remonterait au moins à l’an 1504. Ce terme faisait alors référence aux croyances de l’époque sur l’influence des astres [19], et les véritables causes d’infection étaient inconnues. Une avancée considérable des connaissances sur l’Influenza s’est produite suite aux pandémies de 1781, 1789, 1830, 1847 et 1889. À la fin du 19e siècle, les notions de microbiologie étaient déjà bien établies, mais l’agent pathogène responsable de la grippe n’avait pas encore été isolé. En 1892, les travaux de Pfeiffer et Ki-tasato suggéraient qu’une bactérie, Bacillus Influenzae, était l’agent étiologique d’Influenza. Il est aujourd’hui connu que cette bactérie, maintenant nommée Haemophilus Influenzae, est l’une des causes secondaires de décès liés à Influenza, mais n’en est pas pas l’agent étiologique. C’est en 1898 que le premier virus, celui de la mosaïque du tabac, a été découvert par Martinus Beijerinck. Les virus étaient alors distingués des bactéries principalement par leur capacité à passer au travers de filtres empêchant le passage des bactéries. Au moment de la pandémie de grippe espagnole de 1918, la conception prédominante était encore que Bacillus Influenzae causait l’Influenza. La même année, Olitsky et Gates ont démontré que l’agent causant l’In-fluenza passait au travers des filtres excluant Bacillus Inl’In-fluenzae [13]. En 1933, Wilson Smith, Christopher Andrewes et Patrick Laidlow ont pu isoler le virus causant l’influenza en utilisant une méthode développée par Richard Shope [20].
Durant les années 1940, la distinction entre les Influenzas de type A et B (présentée à la section
1.2.1) a été faite et la production de vaccins a débuté [13]. Le premier antiviral spécifique contre Influenza, l’amantadine, a été approuvé par la « Food and Drug Administration » FDA en 1966. Que ce soit dû au développement de ces outils contre Influenza, ou à des variations intrinsèques dans la virulence des virus, le nombre de décès causés par les pandémies d’Influenza est en diminution depuis 1918 [21].
1.2
Le virus Influenza
1.2.1 Taxonomie
Pour bien cerner les particularités d’Influenza, il est d’abord utile de le situer par rapport aux autres virus en étudiant la taxonomie des virus. En biologie, la taxonomie consiste à catégoriser les organismes selon un ordre hiérarchique. Du plus général au plus spécifique, chaque organisme est attribué à un domaine, royaume, phylum, classe, ordre, famille, genre et espèce [22]. Par exemple, il existe trois domaines biologiques soit les archaea, les bactéries et les eucaryotes.
Les virus sont définis comme des acaryotes (sans noyau) et leur taxonomie débute au niveau des ordres. Ils ne font donc partie d’aucun royaume, phylum ou classe de la biologie. Le comité international sur la taxonomie des virus (ICTV) catégorise actuellement les virus en 6 ordres, mais plusieurs familles de virus, dont celle d’Influenza, ne sont assignées à aucun de ces ordres. Les virus Influenza appartiennent à la famille des Orthomyxoviridae, « myxo » étant un terme grec signifiant mucus [8]. Les virus de cette famille se distinguent entre autres par leur capacité à infecter les vertébrés, leur enveloppe membranaire ou péplos, et leur génome composé de 6 à 8 segments d’ARN à simple brin négatif.
Les Orthomyxoviridae contiennent 5 genres de virus : les isavirus, les thogovirus, ainsi que l’Influenza A, B et C. Il a été proposé d’ajouter un 6egenre d’Orthomyxoviridae pour inclure certains virus non classifiés [23]. Les 3 genres d’Influenza contiennent chacun une seule espèce, ou type, aussi nommés A, B et C. Les virus Influenza de type A, B et C se distinguent les uns des autres par quelques différences dans les protéines codées par leur ARN, tel que présenté à la section suivante.
Les virus Influenza de type A sont reconnus pour présenter une variabilité antigénique plus élevée que les types B et C. Les virus de type A sont donc additionnellement catégorisés par sous-types ou sérotypes selon les variations en acides aminés de leurs protéines de surfaces, soit l’hémagglutinine (HA) et la neuraminidase (NA). Par exemple, un virus contenant des HA de type 5 et des NA de type 1 aura le sérotype H5N1. On compte aujourd’hui 17 HA et 9 NA différentes se distinguant par leur séquence en acides aminés [24, 25]. Il y a 20-74% de variation en acides aminés entre les différents sérotypes d’HA, alors que jusqu’à 9% de variation est observée au sein d’une même sérotype [26]. Seules les HAs de type H1 à H3 et plus rarement H5 se retrouvent chez les souches infectant les humains [24].
Finalement, les virus sont catégorisés par souches. Une souche de virus est définie par son type (A, B ou C), son origine géographique, un numéro attribué lors de son isolation, l’année à laquelle elle a été isolée, et son sérotype dans les cas des virus de type A. Par exemple, la séquence du peptide étudié dans ce projet provient de l’hémagglutinine de type 3 trouvée dans le virus de souche A/Aichi/2/68 (H3N2), aussi retrouvée dans la souche de virus réassorti
X-31.
1.2.2 Structure du virus
Les virus Influenza prennent la forme de sphères d’environ 100 nm de diamètre, ou de filaments d’une longueur d’environ 300 nm [27]. La surface des virus est recouverte de glycoprotéines visibles par micrographie, telle qu’illustrée à la figure1.2.
Figure 1.2: Microphotographie de virus Influenza de type A [28].
La figure 1.3 présente les composantes du virus Influenza de type A. Le matériel génétique viral est contenu au centre du virus sous forme de nucléocapside protéique. La nucléocapside d’influenza, aussi nommée complexe ribonucléoprotéique (RNP), est formée de 8 segments d’ARN, chacun lié à une ARN polymérase trimérique et plusieurs copies de la nucléoprotéine virale (NP) [27]. La nucléocapside est enveloppée d’une matrice protéique composée des pro-téines M1 et M2, et d’une membrane phospholipidique (grand cercle rouge de la figure1.3). Les protéines de surface du virus, l’hémagglutinine (HA) et la neuraminidase (NA), sont ancrées à la membrane.
Le génome d’Influenza A, composé d’environ 13 500 bases d’ARN à simple brin négatif [30], code pour 11 protéines (dont les 3 sous-unités de l’ARN polymérase) nécessaires à la forma-tion du virus. Le génome est physiquement divisé en 8 segments, numérotés selon leur taille et nommés d’après les protéines pour lesquelles ils codent [27, 29]. Les 8 segments sont énu-mérés ci-dessous, en indiquant la position des protéines codées par chacun d’entre eux dans la figure1.3.
– 1) Le segment PB2 code la sous-unité polymérase basique 2 de l’ARN polymérase (PB2, petits triangles gris sur les segments d’ARN).
– 2) Le segment PB1 code la sous-unité polymérase basique 1 de l’ARN polymérase (PB1, petits carrés bleus sur les segments d’ARN) et une protéine proapoptotique (PB1-F2, absente sur le schéma). PB1-F2 est présente chez certaines souches d’Influenza A seule-ment.
Figure 1.3: Schéma du virus Influenza A tiré de [29]. 8 des 11 protéines d’Influenza A sont identifiées par leur acronyme relié à leur structure schématique par une ligne. Les 8 segments d’ARN du virus sont identifiés sous leur structure schématique. Une description détaillée de chaque composante tirée de [27] est donnée dans le texte.
– 3) Le segment PA code la sous-unité polymérase acide de l’ARN polymérase (PA, petits ovales jaunes sur les segments d’ARN). Les protéines PB1, PB2, PA ainsi que NP (codée par le segment 5) se lient à l’ARN viral pour former le RNP.
– 4) Le segment HA code l’hémagglutinine (HA, orange foncé), une protéine de surface virale essentielle à l’entrée du virus dans la cellule hôte. HA est la protéine d’intérêt de ce projet de recherche.
– 5) Le segment NP code la nucléoprotéine (NP, petits cercles rouges sur les segments d’ARN) servant à former le RNP et le nucléocapside.
– 6) Le segment NA code la neuraminidase (NA, orange pâle) nécessaire à la sortie des virus naissants de la cellule hôte.
– 7) Le segment M code les protéines de matrice 1 (M1, ovales gris pâle) et 2 (M2, tubes verts).
– 8) Le segment NS code la protéine non structurale 1 (NS1, absente sur le schéma) et la protéine d’export nucléaire (NEP, absente sur le schéma). NEP est aussi nommée protéine non structurale 2 (NS2). La position des deux protéines non structurales NS1 et NEP dans le virus n’est pas connue.
L’utilité de chaque composante du virus dans le cycle de réplication du virus est détaillée dans la section suivante.
1.2.3 Mécanisme infectieux
Comme tous les virus, Influenza est dépendant des mécanismes de réplication et de trans-cription des cellules eucaryotes pour se répliquer. Le virus doit donc entrer dans une cellule hôte et s’accaparer une partie de la machinerie de la cellule pour produire de nouveaux virus [24, 27,31–33]. Les virus naissants doivent ensuite sortir de la cellule pour pouvoir aller en infecter d’autres. Le cycle de réplication d’Influenza requiert de 4 à 6 heures [8]. La figure
1.4illustre les différentes étapes du cycle infectieux de réplication du virus Influenza. Chaque étape est décrite ci-dessous.
Figure 1.4: Cycle de réplication du virus Influenza. La fusion membranaire, étape d’intérêt de ce projet de recherche, est illustrée en couleurs. Adapté de [34].
a. Adhésion
Une infection Influenza débute par l’adhésion du virus aux cellules hôte. Chez l’humain, les hôtes sont typiquement des cellules épithéliales des voies respiratoires. Les HAs à la surface du virus se lient aux acides sialiques se trouvant sur les glycoprotéines insérées dans la membrane cytoplasmique des hôtes. Les acides sialiques sont attachés aux glycoprotéines de manières différentes selon les espèces, ce qui explique la spécificité des souches d’Influenza pour certaines espèces (aviaire, porcine, humaine ou autre) [27,31]. Les détails de la liaison des HAs à leurs récepteurs glycoprotéiques seront étudiés à la section 1.6.2.
b. Endocytose
L’adhésion du virus déclenche son internalisation par la cellule hôte. L’entrée du virus s’effectue par endocytose grâce à la formation d’une vésicule recouverte de clathrine [31].
c. Fusion membranaire
L’endocytose terminée, la cellule hôte acidifie l’endosome grâce à des pompes à protons. Ceci déclenche une série de changements structuraux des protéines du virus visant à libérer le RNP viral dans le cytoplasme. L’acidification provoque un changement conformationnel de HA qui expose alors le peptide de fusion membranaire (PF), préalablement enfoui dans structure de HA. Le PF s’insère alors dans la membrane endosomale et, avec l’aide d’un second changement structural de HA, fusionne la membrane endosomale avec celle du virus. La fusion membranaire est l’étape du mécanisme infectieux à laquelle s’intéresse ce projet de recherche, et sera détaillée à la section1.5.
d. Dissociation de la nucléocapside et transport du RNP vers le noyau
Les protons présents dans l’endosome traversent aussi jusqu’à la nucléocapside virale grâce au canal ionique M2. L’acidification de l’intérieur du virus provoque un changement de conforma-tion irréversible des protéines M1 qui se dissocient du RNP et le libèrent dans le cytoplasme. Le RNP libéré est importé vers le noyau par des protéines de la cellule hôte, les karyophérines de type importine. Les protéines du RNP viral contiennent des signaux de localisation nucléaire (SLN) qui indiquent aux importines d’importer le RNP dans le noyau. Toutes les protéines du RNP (PB1, PB2, PA et NP) contiennent des SLN. Les SLN sont typiquement de courtes séquences d’acides aminés contenant plusieurs lysines et arginines basiques. Par exemple, la protéine NP contient 3 SLN, dont un contient la séquence KRxxxxxxxxxxxxxRKxR [35].
e. Réplication du virus
Une fois le RNP dans le noyau cellulaire, l’ARN polymérase synthétise l’ARN complémentaire (ARNc) à brin positif à partir de l’ARN viral (ARNv) à brin négatif. L’ARNc est ensuite
utilisé par l’ARN polymérase comme patron pour faire des répliques de l’ARNv. L’ARN poly-mérase synthétise aussi l’ARN messager (ARNm) à brin positif à partir de l’ARNv. L’ARNm provenant du virus est ensuite dirigé vers les ribosomes à l’extérieur du noyau pour être traduit en protéines, comme le serait l’ARNm de l’hôte. Les protéines membranaires (HA, NA et M2) sont synthétisées dans le réticulum endoplasmique rugueux et dirigées vers la membrane cy-toplasmique de l’hôte via l’appareil de Golgi. HA, protéine d’intérêt de ce projet de recherche, nécessite certaines transformations post-transcriptionnelles qui seront détaillées à la section
1.6. Les détails sur le transport des protéines virales non membranaires sont méconnus, mais les protéines du RNP (PA, PB1, PB2 et NP) doivent retourner dans le noyau pour s’associer aux segments d’ARNv nouvellement répliqués. Les RNPs ainsi formés sont exportés hors du noyau par les nucléoporines de l’hôte et la protéine virale NEP [27].
f. Assemblage du virus
Les mécanismes exacts de l’assemblage final des nouveaux virus sont aussi méconnus. Nous savons que l’assemblage a lieu au niveau de la membrane cytoplasmique. Les protéines virales NA, M2 et surtout HA induisent une courbure de la membrane et initient un phénomène de bourgeonnement. Les protéines M1 s’associent ensuite probablement aux parties cytosoliques de HA et NA pour débuter la formation de la nucléocapside tout en augmentant la courbure de la membrane. Les RNPs peuvent à leur tour s’associer aux protéines M1 par l’interaction avec les protéines NP. L’addition graduelle des protéines membranaires virales, de M1 et des RNPs complète le bourgeonnement pour former le virus [36].
g. Relargage du virus
La protéine M2, servant de canal ionique lors de la dissociation de la nucléocapside, joue aussi un rôle durant le relargage du virus. M2 sépare le virus de la membrane cytoplasmique en effectuant une scission membranaire [36]. Le virus reste alors attaché à la cellule par la liaison des HAs aux acides sialiques de l’hôte. NA hydrolyse finalement le lien entre les acides sialiques et les galactoses de glycoprotéines de l’hôte pour relarguer le virus dans le milieu [27].
Fonction des protéines non essentielles
La protéine non structurale NS1 aurait plusieurs fonctions non essentielles qui contribuent à la virulence d’Influenza, telles la suppression de l’immunité et l’inhibition de l’apoptose [37,38]. La protéine NEP, pour « nuclear export protein », sert à l’export des protéines ribonucléiques nouvellement formées dans le noyau de la cellule vers le cytoplasme [39]. Certaines souches d’Influenza A contiennent aussi la protéine PB1-F2, codée sur le segment PB1. La séquence codante de PB1-F2 chevauche celle de PB1 et son cadre de lecture ouvert est déplacé d’une base [27]. PB1-F2 serait proapoptotique en interférant avec le fonctionnement des mitochondries de la cellule hôte [24].
Variations chez les virus de type B et C
La structure virale présentée à la section 1.2.2 et le mécanisme infectieux décrit ci-dessus s’appliquent aux virus Influenza de type A. Les virus de type B et C sont très similaires, mais présentent certaines différences. Chez l’Influenza de type B, le 6esegment d’ARN code pour la protéine NB en plus de NA, et le 7esegment code pour la protéine BM2 au lieu de M2 [40]. NB est un canal ionique non essentiel [40], mais qui augmenterait l’efficacité de réplication virale [41]. BM2 est un canal ionique permettant le passage des protons, comme M2. L’Influenza de type C ne contient que 7 segments d’ARN [42]. L’hémagglutinine de l’Influenza C (HEF), codée sur le 4e segment, remplit le rôle des HA et NA d’Influenza A et B. C’est-à-dire que HEF sert à la fois à l’insertion du virus dans la cellule hôte et à la coupure des acides sialiques lors de la sortie [43]. Le 6e segment de l’Influenza C code la protéine CM2, remplissant aussi la même fonction que M2 et BM2.
1.2.4 Dérive et saut antigénique
Une infection Influenza se termine habituellement après environ une semaine chez les individus en santé. Ceci est dû à l’incapacité des virus à se répliquer suite à la production d’anticorps par le système immunitaire de l’hôte. Les anticorps se lient spécifiquement à certaines régions des protéines de surface du virus pour les empêcher d’accomplir leur fonction. Les protéines NA, et surtout HA, sont les cibles principales d’anticorps. La liaison d’anticorps empêche HA de se lier aux récepteurs de surface de l’hôte ou d’effectuer la fusion membranaire, dépendamment de la région de HA ciblée par les anticorps [44].
Considérant que le système immunitaire conserve une réserve d’anticorps pour les antigènes déjà rencontrés, Influenza ne pourrait pas infecter plusieurs fois un même individu, à moins de pouvoir contourner son immunité. Or, Influenza dispose de tels moyens, soit la dérive et les sauts antigéniques [44].
La dérive antigénique (traduction de antigenic drift ) consiste en l’accumulation de mutations dans les régions liant les anticorps de HA ou NA. Ces mutations rendent les anticorps produits lors d’infections précédentes incapables de se lier aux récentes versions de HA ou NA. Les virus contenant ces mutations sont alors identifiés comme appartenant à une nouvelle souche. Cette nouvelle souche peut alors infecter des individus qui étaient préalablement immunisés, jusqu’à ce que le système immunitaire de ces individus réussisse à produire des anticorps contre les protéines mutées. La dérive antigénique est responsable des épidémies saisonnières d’Influenza. Elle explique aussi pourquoi la composition des vaccins, étudiés à la section 1.3.1, doit être révisée chaque année.
Les sauts antigéniques (traduction de antigenic shift ) ne surviennent qu’avec l’Influenza de type A et consistent en des modifications beaucoup plus brusques que la dérive antigénique. Les sauts surviennent lorsqu’un individu est co-infecté par plusieurs souches différentes d’Influenza
[45]. Les génomes des virus naissants sont alors composés de mélanges des segments des souches infectantes, produisant des virus réassortis drastiquement différents des souches originales. Ces virus réassortis ont le potentiel de causer des pandémies telles que celle observée en 2009, particulièrement si le saut antigénique réintroduit un sérotype d’HA qui était absent des virus circulants pendant une longue période, dans une souche d’Influenza courante [44].
1.3
Contrer l’Influenza
Considérant l’impact d’Influenza sur la santé humaine et les connaissances actuelles sur le virus, certaines interventions ont été développées pour contrer cette infection. Les interventions pharmaceutiques contre Influenza, soit la vaccination et les antiviraux, sont présentées dans cette section. Le présent projet de recherche sur la fusion membranaire d’Influenza pourra avoir un impact sur les futures recherches d’antiviraux, mais n’est aucunement relié à la vaccination. Une attention particulière sera donc portée aux antiviraux basés sur l’inhibition de la fusion membranaire et aux avantages des antiviraux par rapport à la vaccination.
1.3.1 Vaccination
Le principe de la vaccination consiste en l’injection d’une solution contenant des particules virales inactivées ou atténuées afin d’induire la production d’anticorps. Les individus recevant le vaccin deviennent donc immunisés contre la souche virale qu’il contient. Des adjuvants sont souvent ajoutés au vaccin afin de stimuler la production d’anticorps. Typiquement, les vaccins proposés à la population contre la grippe saisonnière sont trivalents et contiennent donc des particules virales inactivées de trois souches, deux d’Influenza de type A et une de type B [46]. À l’inverse, les vaccins utilisés lors de la pandémie telle celle de 2009 sont monovalents et ne contiennent que la souche pandémique.
L’efficacité du vaccin contre Influenza a été démontrée par plusieurs études [47–50]. La vac-cination est donc la première ligne de défense contre l’Influenza. Par exemple, la figure 1.5
compare le nombre d’individus infectés par Influenza en Ontario lors de la pandémie de 2009, avec le nombre projeté dans le cas où il n’y aurait pas eu de vaccination [16]. Le nombre total d’infections a été estimé à environ 3 215 000 (aire sous la courbe bleue de la figure 1.5), et le nombre projeté sans vaccination serait d’environ 4 110 000 (aire sous la courbe rouge de la figure1.5). La vaccination contre la souche pandémique de 2009 aurait donc permis d’éviter 895 000 cas d’Influenza en Ontario seulement, en plus d’empêcher 52 décès.
La vaccination présente cependant certains désavantages. Le premier est que le vaccin ne peut être utilisé en traitement et doit absolument être administré préventivement pour être efficace. Lors d’une pandémie, il est donc nécessaire d’obtenir une grande quantité de vaccins le plus tôt possible afin de limiter l’infection [51]. Lors de la pandémie H1N1 de 2009, la méthode utilisée par GSK pour produire le vaccin ArepanrixMCà partir d’oeufs embryonnés nécessitait
Figure 1.5: Nombre d’infection à l’Influenza H1N1 lors de la pandémie de 2009 (bleu) et nombre projeté d’infections sans vaccination (rouge). Adapté de [16].
environ 6 mois [52]. Ce type de délais de production peut mener à une insuffisance de vaccins, comme celle alors observée aux États-Unis [14]. Une méthode développée par la compagnie Medicago permet une production plus rapide de vaccins grâce à l’expression de particules virales par des plantes transgéniques, et pourrait assurer une meilleure couverture vaccinale lors de prochaines pandémies [53].
Le second problème relié à la vaccination est l’existence d’effets secondaires. La plupart des effets secondaires de la vaccination contre Influenza sont bénins et incluent entre autres des douleurs locales, une légère fièvre et des maux de tête. Des cas de Guillin-Barré, pouvant mener à une paralysie, ont toutefois été rapportés chez 0,002% des vaccinés [15]. La combinaison des effets secondaires des vaccins et de leur utilisation obligatoirement préventive implique d’inoculer des individus sains avec une substance pouvant les incommoder. Considérant la nature relativement bénigne des symptômes d’Influenza, certains individus considèrent que la protection contre Influenza ne surpasse pas les risques des effets secondaires [54]. Il est à noter que l’Université Laval a contribué aux études sur les effets secondaires des vaccins contre la pandémie d’Influenza de 2009. En effet, les résultats des mémoires de Fatoumata Dioubaté [55] et Marie-Claude Gariépy [56], sous la direction du Pr Gaston De Serres, sont en partie inclus dans le Bilan de la direction générale de la santé publique de la vaccination contre la grippe pandémique A(H1N1) 2009 [15].
Le refus de prendre le vaccin par certaines personnes et la difficulté à produire suffisamment de doses rapidement sont responsables du faible taux de vaccination observé lors de la pandémie de 2009. Un haut taux de vaccination avait alors été atteint pour la population canadienne (45%), mais le taux était beaucoup plus bas aux États-Unis (20%), en Australie (25%) et au
Japon (12%) [14]. Ces taux supportent certaines études qui, contrairement à celles citées plus haut, suggèrent que la vaccination contre Influenza sur une population en santé n’apporte que des bénéfices modérés [57,58].
1.3.2 Antiviraux
Les antiviraux sont des molécules se liant spécifiquement à certaines protéines virales pour blo-quer leur fonctionnement et ainsi empêcher la reproduction des virus. Quatre antiviraux ont principalement été utilisés en Amérique du Nord durant les vingt dernières années, soit l’aman-tadine, le rimanl’aman-tadine, l’oseltamivir et le zanamivir. L’amantadine a été approuvé par la FDA (« Food and Drug Administration ») en 1966 sous la marque Symmetrel par Du Pont et Endo Pharmaceuticals [59,60], et est maintenant en vente sous forme générique. Le rimantadine a été approuvé en 1993 et commercialisé sous la marque Flumadine par Forest Pharmaceuticals [61]. L’oseltamivir a été approuvé en 1999 sous la marque Tamiflu par Roche [62]. Le zanamivir a aussi été approuvé par la FDA en 1999, est commercialisé sous la marque Relenza par GSK [63]. L’amantadine [64], l’oseltamivir [65] et le zanamivir [66] sont homologués pour utilisation au Canada, mais le rimantadine ne l’est pas [67].
L’amantadine et le rimantadine sont des inhibiteurs de la protéine de capside virale M2, alors que l’oseltamivir et le zanamivir sont des inhibiteurs de la neuraminidase [1]. Les protéines M2 et neuraminidase sont présentées à la section1.2.2. Les inhibiteurs de la capside virale M2 sont efficaces contre l’Influenza de type A uniquement et les virus y sont devenus largement résistants. Tous les virus de type H1N1 de la pandémie de 2009 sont résistants à l’amantadine et au rimantadine testés selon une étude du « Center for Disease Control and Prevention » [68]. Ces inhibiteurs de M2 sont donc de moins en moins utilisés. L’oseltamivir est aujour-d’hui l’antiviral le plus utilisé contre l’Influenza en Amérique du Nord, suivi par le zanamivir. Le zanamivir est plus efficace que l’oseltamivir sur certaines populations [69]. L’oseltamivir a tout de même pris la principale part du marché [70], probablement parce qu’il est plus facile à administrer. En effet, Tamiflu (oseltamivir) est administré oralement sous forme de capsules, alors que Relenza (zanamivir) est une poudre devant être inhalée oralement à l’aide du Diskhaler®, un inhalateur spécialement conçu par GSK. L’efficacité du zanamivir dépend donc de la capacité respiratoire du patient à qui il est administré [8].
Les antiviraux peuvent être utilisés en traitement ou en prophylaxie (prévention). En traite-ment, ils peuvent être administrés jusqu’à 48 heures après l’apparition des symptômes pour diminuer leur durée de 1 à 2 jours [8]. En prophylaxie, ils sont surtout utilisés pour des indi-vidus ayant été en contact avec des personnes présentant des symptômes d’Influenza.
Les antiviraux présentent toutefois aussi leurs désavantages. Premièrement, la plage de temps où ils peuvent êtres utilisés efficacement comme traitement est relativement courte. 48 heures après l’apparition des symptômes, la prise de l’antiviral par un individu autrement en santé
n’a que peu d’effet sur la durée de l’épisode d’Influenza [8]. On peut supposer qu’après cette période de 48 heures, les virus se sont déjà reproduits en grand nombre et le malade ne peut qu’attendre que son système immunitaire élimine et évacue les particules virales. De plus, tout comme les vaccins, les antiviraux présentent des effets secondaires. L’oseltamivir peut causer des nausées et des vomissements alors que le zanamivir peut causer des difficultés res-piratoires légères et est déconseillé aux asthmatiques [8]. Aucun effet secondaire grave tel le Guillain-Barré n’a cependant été rapporté. Un dernier désavantage majeur des antiviraux est l’apparition de résistances. L’amantadine et le rimantadine sont aujourd’hui peu utilisés pour cause de résistances et certaines souches sont résistantes à l’oseltamivir. Peu de résistances au zanamivir ont été observées à ce jour [69]. Davantage de résistances se développeront néan-moins à l’oseltamivir et au zanamivir à mesure de leur utilisation. Il est à noter des recherches sur la résistance aux antiviraux ont été effectuées à l’Université Laval et sont présentées dans les mémoires de Mariana Baz Etchebarne et Philippe Simon, sous la direction du Pr Guy Boivin [71]. Les conclusions du mémoire de Mariana Baz Etchebarne incluent la citation sui-vante : « ... studies carried out in this master highlight the need of developing new antiviral compounds ... » soulignant l’importance de mieux comprendre les mécanismes infectieux d’In-fluenza comme la fusion membranaire, telle qu’étudiée dans le présent projet de recherche. Puisque les antiviraux sont administrés à des patients souffrant déjà des symptômes d’In-fluenza, ces derniers sont de manière générale très enclins à prendre des antiviraux. 97% des adultes à qui le zanamivir est proposé l’acceptent [8], ce qui est élevé en comparaison des taux de vaccination présentés à la section précédente.
1.3.3 Inhibiteurs de la fusion membranaire
Il existe aussi des antiviraux bloquant la fusion membranaire virale [72]. Le carbobenzoxy-o-Phe-L-PheGly (ZfFG) [73] et la molécule arbidol [74–76] (figure 1.6, gauche) sont reconnus capables d’inhiber la fusion. L’arbidol a été démontré efficace contre Influenza et est aujour-d’hui utilisé en Russie et en Chine. Cependant, cet antiviral n’est pas actuellement utilisé en Amérique du Nord puisqu’il a subi trop peu de tests cliniques [77]. Le mécanisme d’inhibition de la fusion membranaire de l’arbidol est mal connu. Il a été proposé que l’arbidol se lie à la fois à l’hémagglutinine (HA) et aux membranes [76,78]. La figure 1.6illustre l’interaction de l’arbidol avec les phospholipides membranaires selon son insertion mesurée par augmenta-tion de relaxaaugmenta-tion paramagnétique (PRE) [76]. Les phospholipides, parties constituantes des membranes biologiques, seront présentés à la section 1.5.1
L’existence d’inhibiteurs de fusion membranaire confirme qu’il est possible de développer des antiviraux ciblant cette étape du cycle infectieux d’Influenza, et indique que les recherches fondamentales permettant de mieux comprendre la fusion membranaire seront utiles lors de futures recherches d’antiviraux.
Figure 1.6: Structure de l’arbidol (gauche) et interaction proposée avec les phospholipides membranaires (droite). Figures tirées de [76].
1.4
Pharmaco-économie et justification du projet
La section précédente présentait les interventions actuellement disponibles contre Influenza. Il est maintenant important de se demander si ces interventions sont réellement profitables, et donc s’il est justifié d’investir dans la recherche de nouveaux moyens thérapeutiques contre Influenza. Après tout, le taux de fatalité d’Influenza semble relativement bas dans le contexte actuel, considérant le peu de décès enregistrés lors de la pandémie H1N1 de 2009 (voir section
1.1.2). Aussi, serait-il plus avantageux d’investir sur la vaccination ou sur les antiviraux ? Afin de trancher sur ces questions, la dimension économique doit être considérée, particulièrement pour une infection comme Influenza, qui ne présente pas un haut taux de mortalité, mais qui emmène chaque année des pertes économiques importantes (voir section1.1.1).
La économie est le domaine s’intéressant à la rentabilité des interventions pharmaco-logiques. Cette section présente un résumé des études de pharmaco-économie sur la vaccination et les antiviraux contre Influenza. L’objectif est de justifier ce projet en démontrant que les interventions antivirales sont profitables. Rappelons que le présent projet de recherche ne vise pas directement le développement de nouveaux antiviraux, mais que les informations fonda-mentales sur la fusion membranaire qui y seront obtenues pourront êtres utilisées dans les futures recherches d’antiviraux.
1.4.1 Exemple de la campagne de vaccination H1N1 de 2009
Il est intéressant de commencer l’étude pharmaco-économie de la vaccination avec un exemple récent. Une campagne de vaccination à grande échelle a eu lieu au Canada en 2009 pour conte-nir la pandémie d’Influenza H1N1. Environ 45% de la population canadienne a été vaccinée [15]. Le Canada a commandé 50 millions de doses de vaccins, soit pour vacciner 75% de la po-pulation à deux doses par personne. Les vaccins ArepanrixMC ont été achetés de la compagnie
GlaxoSmithKline (GSK) à 8$ la dose pour un total de 400 millions $ [16]. Les vaccins ont été produits dans l’usine de GSK située à Québec [79]. GSK a aussi fait un don de 60 millions de doses à l’OMS pour fournir les pays à bas revenu [80]. Le coût total de l’opération de vacci-nation de masse canadienne, incluant l’achat, la distribution et la promotion des vaccins est évalué à 20-30 $ la dose, soit 1-1,5 milliards $ [16,81]. Ceci est une somme considérable, mais tout de même bien moindre que les frais de 9 milliards $ causés par l’Influenza saisonnière, estimés à la section1.1.2.
1.4.2 Rapport coût-efficacité différentiel
L’exemple précédent illustre que plusieurs facteurs sont à prendre en compte pour évaluer la rentabilité d’une intervention contre Influenza. Le rapport coût-efficacité différentiel (RCED), traduit de l’anglais « incremental cost-effectiveness ratio » (ICER), tente d’intégrer tous ces facteurs en une valeur significative. Le RCED est le coût total de l’intervention pour le système de santé (tel que décrit au paragraphe précédent) divisé par le nombre d’années de vie gagnées ajustées par la qualité de vie (AVAQ), traduit de l’anglais « quality adjusted life-year » (QALY) [8, 82]. L’AVAQ est calculé comme le nombre total d’années gagnées par les individus de la société où l’intervention pharmacologique a lieu, pondéré par la qualité de vie des individus [83]. Par exemple, une intervention sauvant la vie d’un jeune adulte fera gagner plus d’AVAQ qu’une intervention sauvant la vie d’un individu dont l’âge approche ou dépasse l’espérance de vie. Aussi, une intervention permettant d’éviter des séquelles à un individu présentera un gain d’AVAQ en évitant une perte de qualité de vie de l’individu pour le reste de sa vie. Bien sûr, la qualité de vie d’un individu ne dépend pas seulement de son âge et état de santé, et sa quantification peut être erronée. La pondération des années incluses dans l’AVAQ est un domaine d’étude en soi [83]. Ceci étant dit, le RCED est un excellent outil pour évaluer la rentabilité d’une intervention pharmacologique. Selon l’OMS, une intervention est considérée comme hautement rentable si son RCED ne dépasse pas le PIB par habitant de la société à laquelle l’intervention est appliquée, et rentable si le RCED ne dépasse pas trois fois le PIB [84, 85]. Autrement dit, les dépenses encourues par l’intervention sont alors compensées par le gain de productivité de la population. Au Canada, une intervention est donc considérée hautement rentable si son RCED est en dessous d’environ 40 000$ [16]. Le RCED peut alors être utilisé pour comparer quantitativement les différentes interventions contre Influenza. L’utilisation du RCED soulève des questions éthiques intéressantes. Cet outil pharmaco-économiques implique d’attribuer une valeur monétaire à la vie humaine, ce qui peut brusquer le sens éthique de certains. Le fait que le soutien du métabolisme d’un être vivant requiert une certaine quantité de ressources est malgré tout inévitable, et ce, depuis plusieurs milliards d’années. À l’échelle évolutive, cela fait très peu de temps que l’humain a un pouvoir sur la survie des individus malades. À mesure que nos techniques médicales avancent, nous pouvons préserver la vie d’individus souffrant de conditions de plus en plus graves, en utilisant des
ressources de plus en plus importantes. La stratégie visant à sauvegarder la vie de tout indi-vidu à tout prix risque de drainer une énorme portion des ressources disponibles, diminuant la qualité de vie de la population en général au lieu de l’améliorer. Le décès des individus, qui était auparavant du domaine de l’incontrôlable, deviendra éventuellement un choix conscient, et l’est déjà partiellement.
1.4.3 Méta-analyses
Un grand nombre d’études sur la rentabilité des interventions contre Influenza ont été effectuées en utilisant le RCED ou des outils semblables [16,49–51,54,58,86–88]. Considérant la diffi-culté d’évaluer l’efficacité d’une intervention et des paramètres comme l’AVAQ, les conclusions de ces études sont souvent contradictoires. Les antiviraux sont parfois trouvés plus rentables que la vaccination [86], ou l’inverse [54]. Certains auteurs ont donc entrepris de rassembler ces études pour faire une méta-analyse et ressortir les tendances clés [1,8,89].
Les RCED rapportés à la table1.1proviennent de deux méta-analyses effectuées sur l’Influenza saisonnière [1,8]. Puisque ces études ont été publiées dans un journal britannique, les valeurs monétaires y sont inscrites en livre sterling (£) et sont ici converties en dollars canadiens. Les valeurs monétaires ont été converties en dollars canadiens de 2012 en convertissant d’abord d’après le taux de change de 2003 (année où la méta-analyse a été effectuée [8]), soit 2,288 [90], et en incluant ensuite l’inflation de 17,9% depuis 2003 [91]. La résistance de certaines souches virales à l’amantadine n’a pas été considérée dans ces études [8].
Table 1.1: Rentabilité des interventions contre Influenza selon leur rapport coût-efficacité différentiel (RCED) en $/AVAQ. Un RCED faible dénote une intervention rentable. Les valeurs ont été converties de £ de 2003 en $ canadiens de 2012. Les valeurs pour la vaccination proviennent du modèle de base de Turner et coll. [8] Les valeurs pour les anti-viraux proviennent du modèle extrapolé de Turner et coll. rapporté par Burch et coll. [1] p. 86.
Population Vaccination Amantadine Zamamivir Oseltamivir En santé 27 475 16 700 23 970 12 758 À risque 6 295 12 235 8 172 8 137
Les valeurs de RCED de la table 1.1 se trouvent entre 6 295 et 27 475 $/AVAQ. Ceci ne correspond pas à un coût par intervention, mais bien au coût moyen par individu recevant le traitement, divisé par le nombre moyen d’AVAQ gagnés par ces individus. Dans le cas de l’Influenza, le coût du traitement est relativement faible, mais le gain en AVAQ l’est aussi. Basé sur les prix de l’année 2000 [92], le prix par traitement de ces antiviraux est d’environ 56$ pour Zanamivir, 70$ pour Oseltamivir, 34$ pour Rimantadine et 7$ pour Amantadine. Le prix des vaccins monovalents utilisés lors de la pandémie de 2009 était de 8$ la dose [16]. La perte en AVAQ d’un épisode d’Influenza est estimé à 0,02 [93] et les interventions pharmaceutiques ne permettent de regagner qu’une fraction de ces AVAQ. Ainsi, même si les
coûts des interventions contre Influenza sont bas, leurs ICER peuvent être comparables à ceux d’interventions beaucoup plus coûteuses. Par exemple, un traitement coûtant 60 000$, mais augmentant de 3 ans l’espérance de vie d’un individu de la population active, aurait un ICER de 20 000 $/AVAQ.
Les résultats de la table 1.1suggèrent que toutes les interventions contre Influenza présentées sont hautement rentables au Canada, car aucune n’a de RCED dépassant le PIB par habi-tant. Pour la population en santé, les antiviraux ont un RCED plus bas que la vaccination et seraient donc plus rentables que la vaccination. À l’inverse, le RCED de la vaccination est plus bas que celui des antiviraux pour la population à risque. Ceci est logique, puisque vacciner une grande proportion de la population en santé implique nécessairement de vacciner beaucoup de gens qui n’auraient pas été malades. Les résultats de méta-analyses indiquent que les antiviraux sont avantageux par rapport à la vaccination pour la population en santé, supportant l’importance de développer de nouveaux antiviraux. Combiné à la possibilité de produire des inhibiteurs de fusion membranaire démontrée à la section 1.3.3, ceci motive ce projet de recherche fondamentale sur le mécanisme de fusion membranaire du PF d’Influenza.
1.5
La fusion membranaire
Maintenant que la justification du projet a été exposée, les connaissances actuelles sur le méca-nisme de fusion servant de base au projet peuvent être présentées. Cette section décrit la fusion membranaire en détail, en commençant par présenter les membranes et les phospholipides qui les composent.
1.5.1 Membranes
Phospholipides
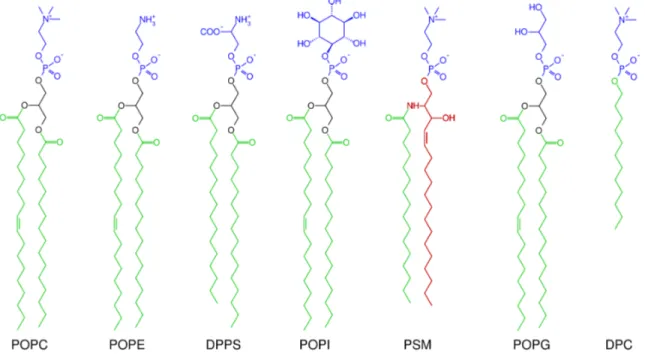
Les principaux constituants des membranes cellulaires sont les phospholipides. Les phospholi-pides sont des molécules amphiphiles composées d’acides gras et de groupements phosphates, souvent reliés à un alcool polaire [94,95]. Sauf pour certaines exceptions, les acides gras sont reliés au phosphate par un glycérol. La figure 1.7 présente la structure de certains phospho-lipides importants dans la constitution des membranes eucaryotes, bactériennes et modèles. La nomenclature des phospholipides se base sur la position des groupements par rapport au glycérol (noir dans la figure 1.7). Dans le 1-palmitoyl-2-oléoyl-3-sn-phosphatidylcholine par exemple, la position 1 du glycérol est estérifiée avec un acide palmitique, la position 2 avec un acide oléique et la position 3 avec une phosphatidylcholine. La notation sn signifie que la nomenclature du composé suit la numérotation stéréochimique, établie afin de distinguer les différents stéréoisomères [96]. Puisque tous les phospholipides contenant un glycérol présen-tés ci-dessous sont estérifiés avec un acide gras en position 1 et 2, et liés à un phosphate en position 3, une nomenclature simplifiée est utilisée. Ainsi, la figure 1.7présente le
palmitoyl-oléoyl-phosphatidylcholine (POPC), le palmitoyl-oléoyl-phosphatidyléthanolamine (POPE), le dipalmitoyl-phoshatidylsérine (DPPS), le palmitoyl-oléoyl-phosphatidylinositol (POPI), le palmitoyl-sphingomyéline (PSM), le palmitoyl-oléoyl-phosphatidylglycérol (POPG) et dodé-cylphosphatidylcholine (DPC).
Figure 1.7: Structure de phospholipides d’intérêt. Têtes polaires incluant les phosphates en bleu. Glycérols en noir. Acides gras en vert. Pour le PSM, la sphingosine remplaçant le glycérol est en rouge. La tête polaire du PSM peut être une choline, telle qu’illustrée, ou une éthanolamine. Image construite à l’aide du logiciel libre BKChem [97].
Dans les cellules épithéliales, cibles du virus Influenza, les phospholipides contiennent des têtes polaires de phosphatidylcholine (PC) (∼ 50 %), sphingomyéline (SM) (∼ 20 %), phosphati-dyléthanolamine (PE) (∼ 15 %), phosphatidylinositol (PI) (∼ 10 %) et phosphatidylsérine (PS) (∼ 5 %). Les têtes polaires de PI et PS sont chargées négativement, alors que celles de PC, PE et SM sont zwitterioniques. Les acides gras auxquelles ces têtes sont reliées sont principalement les acides oléiques avec 18 carbones et une insaturation au 9e carbone (∼ 26 %) ou au 7e carbone (∼ 7 %), l’acide palmitique avec 16 carbones saturés (∼ 23 %), l’acide stéarique avec 18 carbones saturés (∼ 15 %) et les acides palmitoléiques avec 16 carbones et une insaturation au 7e carbone (∼ 5 %) ou au 9e carbone (∼ 3 %) [98,99].
Les phospholipides POPC, POPE, DPPS, POPI et PSM, illustrés à la figure1.7, sont parmi les principaux phospholipides retrouvés dans les membranes épithéliales [100] et les membranes des cellules hôtes d’Influenza [98]. Les phospholipides POPG et DPC sont quant à eux utilisés dans les membranes modèles lors d’expériences in vitro. Une composition lipidique souvent utilisée pour modéliser les membranes cellulaires eucaryotes est celle contenant des POPCs et des POPGs dans des proportions de 4 pour 1. En effet, les POPGs possèdent une charge électrostatique totale négative, tout comme les phospholipides ayant des têtes de PI ou PS qui composent ensemble environ 15 % des phospholipides membranaires [101]. Il est toutefois à noter que les POPGs ne se retrouvent habituellement pas dans les membranes de cellules eucaryotes, mais plutôt dans celles des bactéries [102].
Figure 1.8: Phases lipidiques. Les têtes polaires sont représentées par des cercles et les chaînes grasses par des traits ondulés. Tiré de [103].
Phases lipidiques
Les phospholipides ont un caractère amphiphile, présentant une partie hydrophile (tête po-laire) et une partie hydrophobe (chaînes grasses). En solution aqueuse, les phospholipides s’as-semblent spontanément pour regrouper leurs parties hydrophobes et minimiser les contacts des chaînes grasses avec l’eau. Cet assemblage forme différentes structures, appelées phases lipidiques. Les principales phases lipidiques sont les phases lamellaire, hexagonale, micellaire et cubique, illustrées à la figure 1.8. Les membranes, soit les bicouches phospholipidiques, cor-respondent à la phase lamellaire. En phase hexagonale, les chaînes grasses se rassemblent au centre de structures en tubes, entourées des têtes polaires et du solvant aqueux. Dans la phase micellaire, les phospholipides forment des sphères, aussi avec les chaînes grasses vers le centre et les têtes polaires pointant vers l’extérieur de la sphère. La phase cubique permet différentes structures périodiques relativement complexes [103]. À concentration phospholipidique élevée, les phases hexagonales et micellaires s’inversent, séquestrant le solvant aqueux au centre des structures et dirigeant les chaînes grasses vers l’extérieur des structures. La phase d’une solu-tion lipidique dépend de plusieurs facteurs, dont la longueur des chaînes lipidiques, la taille de
![Figure 1.3: Schéma du virus Influenza A tiré de [29]. 8 des 11 protéines d’Influenza A sont identifiées par leur acronyme relié à leur structure schématique par une ligne](https://thumb-eu.123doks.com/thumbv2/123doknet/6719126.184883/29.918.222.743.125.591/schéma-influenza-protéines-influenza-identifiées-acronyme-structure-schématique.webp)
![Figure 1.11: Structures de membranes artificielles utilisées dans l’étude expérimentale et théorique des membranes [115].](https://thumb-eu.123doks.com/thumbv2/123doknet/6719126.184883/46.918.162.702.275.774/figure-structures-membranes-artificielles-utilisées-expérimentale-théorique-membranes.webp)
![Figure 1.12: Densité des différentes régions des bicouches lipidiques. Tiré de [128].](https://thumb-eu.123doks.com/thumbv2/123doknet/6719126.184883/48.918.264.611.105.618/figure-densité-régions-bicouches-lipidiques-tiré.webp)
![Figure 1.13: Profil de pression latérale des bicouches. Adapté de [130].](https://thumb-eu.123doks.com/thumbv2/123doknet/6719126.184883/49.918.172.804.129.311/figure-profil-pression-latérale-bicouches-adapté.webp)
![Figure 1.18: Changement conformationnel pH-dépendant de l’hémagglutinine [162, 163]. À gauche, structure cristallographique de HA à pH neutre tirée du PDB 1HGE [146]](https://thumb-eu.123doks.com/thumbv2/123doknet/6719126.184883/56.918.181.713.121.449/figure-changement-conformationnel-dépendant-hémagglutinine-gauche-structure-cristallographique.webp)
![Figure 1.21: Structures alternatives du PF. Structure avec charnière flexible obtenue par RMN en solution de micelles [191] (gauche)](https://thumb-eu.123doks.com/thumbv2/123doknet/6719126.184883/60.918.131.760.412.579/figure-structures-alternatives-structure-charnière-flexible-solution-micelles.webp)
![Figure 1.24: Schéma illustrant l’effet du peptide sur les membranes phospholipidiques [186].](https://thumb-eu.123doks.com/thumbv2/123doknet/6719126.184883/62.918.276.599.834.1003/figure-schéma-illustrant-l-effet-peptide-membranes-phospholipidiques.webp)


