A curve-crossing model to rationalize and optimize diarylethene dyads
Texte intégral
Figure
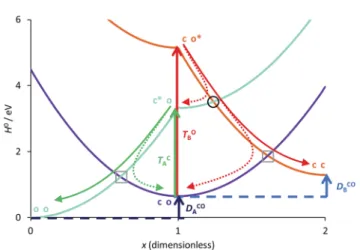
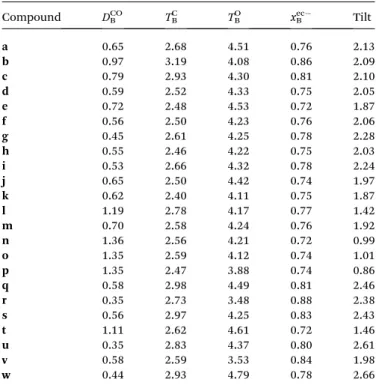
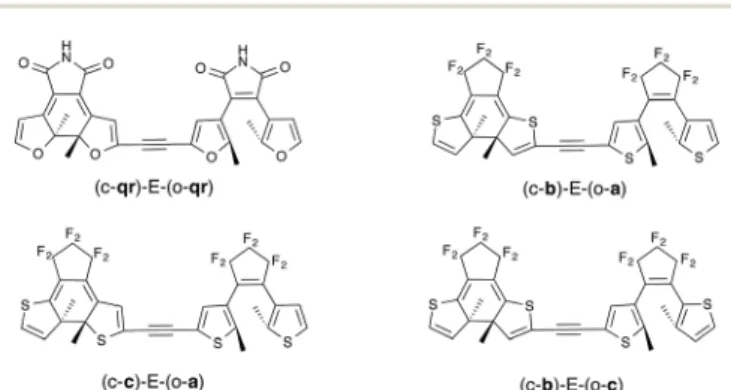
Documents relatifs
For a space curve in pseudo-generic position, the connec- tions between real singularities and regular points are ex- actly those obtained on the projected curve using
Lehmer conjecture, Lehmer problem, Lehmer’s number, Schinzel-Zassenhaus conjecture, mapping class, stretch factor, coxeter system, Salem number, Perron number, Pisot number,
The fundamental group of U (whose universal cover is H d )@ ’TT’ 1 (U), classifies therefore phonon topological (dislocations of Burgers’ vectors b equal to a
For a given parametric representation of a curve, PTOPO computes the special points on the curve, the characteristic box, the corresponding graph, and then it visualizes the
Vous pouvez aussi rejoindre l’un des nombreux GROUPES associés, ou le quitter si un ami vous y a inclus et qu’il ne vous convient pas (beaucoup l’ignorent, mais il est
Dans notre étude les patients étaient répartis selon la sévérité de l’hémophilie en 52,59% hémophiles sévères, ce qui est proche de la littérature qui détermine
Given an energy functional or hamiltonian Je, one metastable configuration from each class may be found by minimising within that class. The topo- logical
Generally speaking, the topological obstructions connected to the existence of linear defects, live in the fundamental group of the manifold of internal states ; in the
