Université de Montréal
Lipoprotein-Associated Phospholipase A
2(Lp-PLA
2) in
Acute Coronary Syndrome
par Bashar Jabor
Programme de Sciences Biomédicales Faculté de Médecine
Mémoire présenté à la Faculté de Médecine en vue de l’obtention du grade de Maîtrise
en Sciences Biomédicales option Générale
Décembre 2014
Abstract
Lipoprotein associated phospholipase A2 (Lp-PLA2) is a biomarker of several inflammatory diseases and syndromes. An elevated Lp-PLA2 level is associated with unstable atherosclerotic plaques. Bound to plasma lipoproteins (LDL and HDL), Lp-PLA2 prevents the formation of biologically active oxidized phospholipids on their surface such as oxidized phosphatidylcholine (oxPC). Nevertheless, the products of Lp-PLA2 action, lysophosphatidylcholine (LPC) and non-esterified fatty acids (NEFA) are both known to aggravate inflammation.
Thus, understanding the metabolism of Lp-PLA2 could help us better understand its role in plaque formation, as studies have shown high expression of Lp-PLA2 and LPCs in unstable plaques. Moreover, studies showed correlation between increased Lp-PLA2 mass and activity and increased risk of coronary artery disease, stroke, and death. The inhibition of Lp-PLA2 with a small molecule, Darapladib, has not demonstrated benefit in reduction of cardiovascular events in two clinical studies.
Here, the first chapter will focus on Lp-PLA2 and cardiovascular disease in man, highlighting the latest updates in the literature. The second and third chapters will introduce experimental work on Lp-PLA2 in the setting of acute coronary syndrome.
Keywords : Lp-PLA2, Lipoprotein-associated phospholipase A2, PAF-AH, platelet activating factor acetyl hydrolase, acute coronary syndrome, ACS, cardiovascular diseases, HDL, LDL
Résumé
La phospholipase A2 liée aux lipoprotéines (Lp-PLA2) est une biomarqueur de plusieurs maladies inflammatoires et une niveau sérique élevé est associé à l’instabilité de la plaque artérioscléreuse. Comme son nom l’indique, la Lp-PLA2 est liée aux lipoprotéines plasmatiques (LDL et HDL) et son rôle est de prévenir l’accumulation de phospholipides oxidés a la surface des lipoprotéines. Toutefois, les produits de dégradation des phospholipides oxidés par la Lp-PLA2 - le lysophosphatidyl choline par les acides gras oxidés peuvent aussi promouvoir l’inflammation.
Mieux comprendre le métabolisme de la Lp-PLA2 pourrait nous permettre de mieux apprécier son rôle dans la formation d’une plaque artérioscléreuse instable, car des études antérieures ont démontré une forte expression de la Lp-PLA2 dans la plaque. De plus, il existe une forte corrélation entre les niveaux et l’activité plasmatiques de la Lp-PLA2 et la maladie coronarienne, les accidents cérébraux-vasculaires et la mortalité cardiaque. L’inhibition de la Lp-PLA2 avec une petite molécule, le darapladib, n’a pas démontré de bénéfice sur les évènements cardiovasculaires dans deux études cliniques.
Cette thèse présentera d’abord une revue de la littérature sur la Lp-PLA2 et les maladies cardiovasculaires et les deuxième et troisième chapitres, une étude clinique réalisée sur des patients avec un syndrome coronarien aigu.
Mots-clés : Phospholipase A2 liée aux lipoprotéines (Lp-PLA2); Facteur d’activation des plaquettes (PAF); acyl hydrolase (AH); syndrome coronarien aigu (ACS); lipoprotéines de haute densité (HDL); lipoprotéines de basse densité (LDL).
Table of Contents
Abstract... i
Résumé... ii
Table of contents... iii
List of tables... v
List of figures... vi
Acknowledgements... vii
List of abbreviations... viii
Introduction Chapter I: Lp-PLA2: a brief introduction... 1
1.1 Introduction... 2
1.2 Biochemistry of Lp-PLA2... 5
1.2.1 Structure of Lp-PLA2... 5
1.2.2 Lp-PLA2 and PAF... 6
1.2.3 Substrate specificity... 8
1.2.4 Products of Lp-PLA2 and their biologic effects... 9
1.2.5 Regulation of Lp-PLA2 ... 10
1.3 Lp-PLA2 animal models... 12
1.4 Lp-PLA2 and statins... 13
1.5 Controversy of Lp-PLA2 function... 13
1.6 Epidemiologic studies of Lp-PLA2... 15
Clinical research
Chapter II: Hypothesis, aims and objectives... 20
Chapter III: Lipoprotein-Associated Phospholipase A2 (Lp-PLA2) in Acute Coronary Syndrome: Relationship With Low-Density Lipoprotein Cholesterol... 22
Abstract... 23 Résumé... 25 Methods... 30 Results... 33 Discussion... 43 References... 46
Chapter IV: Discussion, conclusion and future work... 49
4.1 Discussion, conclusion... 50
4.2 Future work... 55
References... 56
List of tables
Table 1. Study subject characteristics...34 Table 2. Pearson correlation coefficients of Lp-PLA2 mass with cardiovascular risk
List of figures
Chapter I
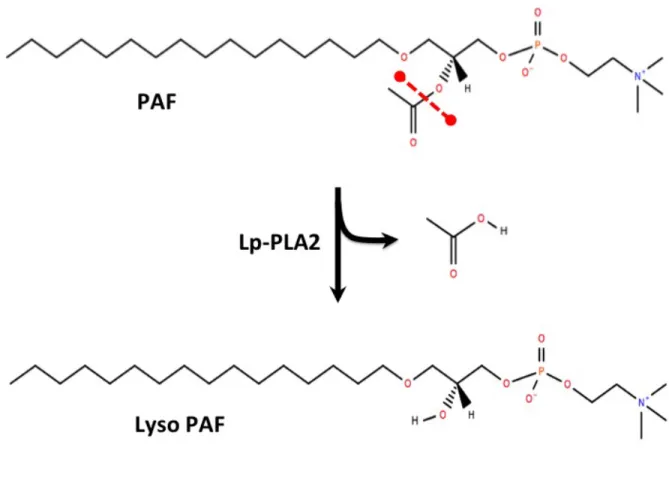
Figure 1. Hydrolysis of PAF by Lp-PLA2 (PAF-AH)... 7
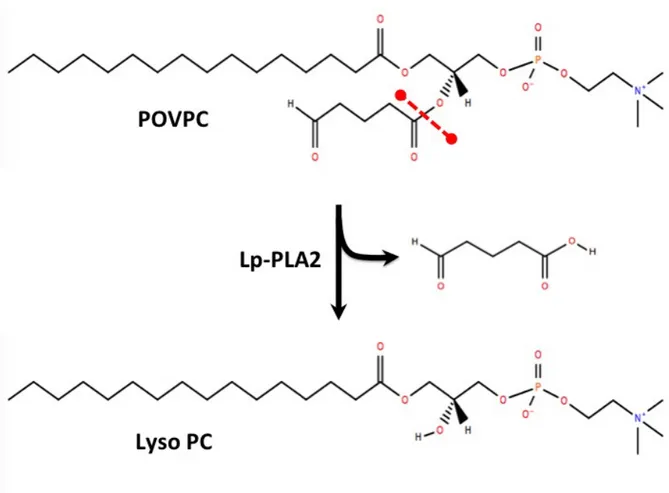
Figure 2. Hydrolysis of an oxidized phospholipid (POVPC) by Lp-PLA2... 8
Chapter III
Figure 1. hsCRP and HDL-C (inset) levels in study subjects... 35 Figure 2. (A) Lp-PLA2 levels in the plasma of study subjects... 36
(B) Difference in Lp-PLA2 mass between ACS acute and recovery (C) Difference in LDL-C between the same groups
Figure 3. Distribution of Lp-PLA2 mass... 39
Figure 4. Correlations between Lp-PLA2 and LDL-C... 41
Annex 1
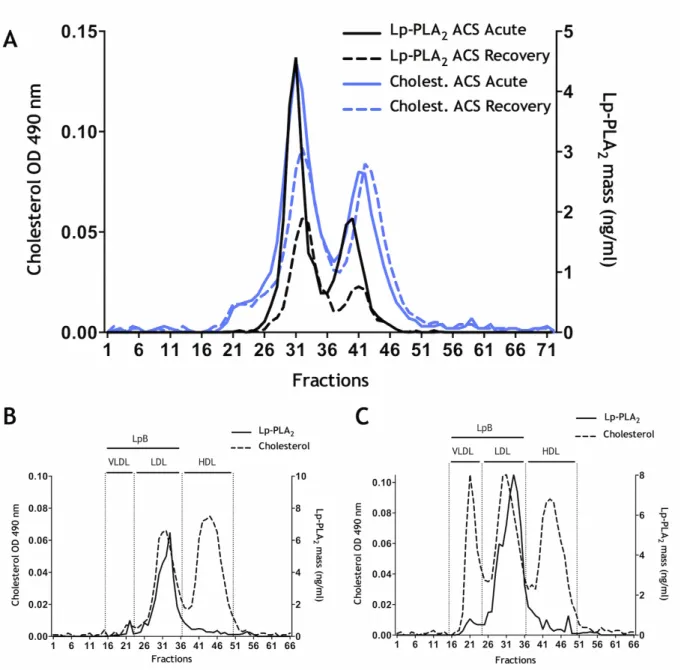
Supplementary Figure 1. (A) Lp-PLA2 mass in the apoB-containing lipoprotein fraction (LpB) obtained by UTC...65
Acknowledgements
I would like to start by showing my deep appreciation to my supervisor Dr. Walid Mourad for his guidance, patience, and enlightenment that enriched my graduate experience.
I would also like to express my gratitude to Dr. Adel Giaid and Dr. Daniel Lajeunesse for always receiving me with open arms, and for their prudent advice and support throughout my studies.
Great appreciation goes to my colleagues and friends Hong and Isabelle for their equal efforts in this study. I also thank my friends Steve and Leo for their scientific advice. I would also like to thank our beloved Colette Rondeau, RN, for her help in recruiting patients for this study.
And finally, my deepest gratitude goes to my co-supervisor and mentor Dr. Jacques Genest. His high expectations and exceptional leadership provided me with pillars of success, both as a master’s student and a physician. Dr. Genest is a role model for clinicians and scientists, and is indeed a true gentleman.
List of abbreviations
ACS: acute coronary syndrome Apo A1: apolipoprotein A1 Apo B: apolipoprotein BCAD: stable coronary artery diseases CVD: cardiovascular diseases
HDL: high density lipoprotein
HDL-C: high-density lipoprotein cholesterol HPLC: high-performance liquid chromatography hsCRP: high-sensitivity C-reactive protein LDL: low density lipoprotein
LDL-C: low-density lipoprotein cholesterol LPA: lysophosphatidic acid
LpB: apoB-containing lipoprotein fraction LPC: lysophosphatidylcholine
Lp-PLA2: Lipoprotein-associated phospholipase A2 NEFA = non-esterified fatty acids
oxPC = oxidized phosphatidylcholine
PAF = platelet activating factor (1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine) PAF-AH = platelet activating factor acetyl hydrolase
PLA2 = Phospholipase A2 UTC: ultracentrifugation
Chapter I: Introduction
Lipoprotein-Associated Phospholipase A
2(Lp-PLA
2): a brief
1.1 Introduction
Cardiovascular diseases are the leading cause of death worldwide. Over decades, studies aimed to identify and evaluate cardiovascular risk in individuals. The Framingham Heart Study is probably the frontrunner in this field. Launched in 1948, Framingham’s first cohort included more than 5,000 participants, and followed in recruiting around 9,000 subjects from their offspring in a second and a third cohorts (1971 and 2002) for a total of more than 14,000 participants. Today, our clinical guidelines include “traditional” modifiable risk factors for cardiovascular diseases: smoking, high cholesterol levels, diabetes, hypertension, obesity, low physical activity and unhealthy diet. Targeting those risk factors by clinicians has resulted in a 50% reduction in age adjusted mortality rates in the US over a 50-year period. In Canada, we see a glimpse of progress as cardiovascular diseases (for the first time) ranked second instead of first as a leading cause of death in 2008. This has largely been confirmed in many observational studies, especially the INTERHEART study. 1-4 However, an observational study by the National Registry of Myocardial Infarction (NRMI) showed that out of a total of 542,008 patients presented with a first acute myocardial infarction between 1994 and 2006, an overall 78,103 (14.4%) had none of the risk factors mentioned above, hence the need for additional biomarkers of cardiovascular risk. 5
Coronary artery disease (CAD) is overwhelmingly caused by atherosclerosis, a process that involves long-term chronic inflammation of the arteries. Thus, the relationship between inflammation, atherosclerosis and the high incidence of cardiovascular diseases (CVD) has been extensively studied. The inflammatory process is triggered by a combination of lipid accumulation in the arterial wall that might undergo oxidation over time (oxLDL), and
activation of several types of inflammatory cells (macrophages, T-cells, mast cells). That results in the formation of arterial plaques that may or may not be stable. Foam cells are lipid-laden macrophages that undergo apoptosis and form the necrotic core of the arterial plaque. In susceptible plaques, the production of certain cytokines e.g. interferon-g (IFN-g) diminishes collagen synthesis and results in plaques with thin fibrous caps. Moreover, the production of proteases by inflammatory cells can disrupt the plaque’s fibrous cap leading to plaque rupture. A series of events characterized by platelet activation and thrombus formation follow, and may lead to arterial occlusion and a subsequent event of acute coronary syndrome (ACS) (such as myocardial infarction). On the other hand, stable plaques (less prone to rupture) continue to grow and cause reduction in the arterial blood flow, and can manifest as stable ischemic heart disease. 6-8
With the need for new biomarkers for CVD and atherosclerosis, research has yielded new potential biomarkers such as high-sensitivity C-reactive protein (hsCRP), Brain natriuretic peptide (BNP), anti-phosphorylcholine (Anti-PC), and Lp-PLA2. 9
Lp-PLA2, also known as platelet activating factor acetyl hydrolase (PAF-AH), is a member of the phospholipase A2 (PLA2) family, group VIIA. According to the biochemical structure and properties of its members, Phospholipase A2 family is classified into 5 categories (secreted PLA2, cytosolic PLA2, Ca+ independent PLA2, lysosomal PLA2, and PAF-AH PLA2) divided in 15 groups. PLA2 enzymes hydrolyze the ester bond at the sn-2 position of phospholipids producing free fatty acids (FFA) and lysophospholipids. 10
In the arterial wall, several phospholipids on LDL undergo oxidation altering their structure to yield oxLDL, which can be taken up by macrophages. OxLDL molecules contain different bioactive compounds that may include phospholipid products, sphingolipid products, free fatty
acid products, oxysterols, cholesteryl ester products, hydroxynonenal and malondialdehyde, and products of apolipoprotein B (ApoB) modification. 11
During the oxidation process, reactive oxygen species attack fatty acyl chains and cause them to undergo peroxidation to generate peroxides that provoke different signalling pathways that end up aggravating the inflammation process. 12 Unsaturated chains are more prone to oxidation than saturated fatty acids. Moreover, short-medium chained fatty acids are more prone to oxidation than long chained fatty acids. 13, 14 Therefore, polyunsaturated fatty acids (PUFA) commonly located on the sn2 position of the fatty acyl chain of the phospholipid are easier targets for oxidation due to the weak hydrogen-carbon bonds in methylene groups, and are believed to be the main source of various oxidized phospholipids. 12, 15, 16
While PLA1 enzymes cleave fatty acyl chains at the sn1 position, oxidized phospholipids at the sn2 position trigger Lp-PLA2, yielding two main products: lysophosphatidylcholine (LPC) and nonesterified fatty acids (NEFA). 14
1.2 Biochemistry of Lp-PLA2
1.2.1 Structure of Lp-PLA2
Lp-PLA2 is a 45 KDa protein that consists of consists of 441 amino acids and doesn’t require Ca+ to perform its function. The crystal structure of Lp-PLA2 has been identified. 17 The catalytic site consists of a triad of serine, aspartate, and histidine, located on residues 273, 296, and 351 respectively, characterized Lp-PLA2 in a class separate from other PLA2 enzymes that lack the serine. 10
Various cells secrete Lp-PLA2 such as macrophages, Kupffer cells, platelets, and mast cells, although it is likely that macrophages have the biggest role. Once Lp-PLA2 is secreted into the plasma, it binds to lipoproteins (mainly HDL and LDL) and circulates as an active enzyme. Some studies reported Lp-PLA2 distribution as approximately 80% on LDL and 20% on HDL, while others reported that it is around two-thirds on LDL vs. one third on HDL, and that Lp-PLA2 mass in the plasma is more dependent on LDL-C levels. 18, 19 The interaction between Lp-PLA2 and LDL occurs between the N-terminus part of Lp-PLA2 and the C-terminus part of ApoB. 16 A previous study has shown that residue 205 on Lp-PLA2 is involved in that binding. On the other hand, it seems that Lp-PLA2 binds to HDL at three different sites, residues 113– 120, 192–204, 360–368, with 192–204 being the region where Lp-PLA2 binds to apolipoprotein A1 (ApoA1). 20
Previous work has shown that Lp-PLA2 is associated with Lipoprotein a (Lp(a)) in the plasma, 21, 22 while other studies have demonstrated the presence of a higher tendency of binding between Lp-PLA2 and small dense electronegative LDL subfraction (LDL-5) and very high-density lipoprotein-1 subfraction (VHDL-1) in human plasma. 16, 23 A genome-wide association study by Thanassoulis et al recently discovered an association between Lp(a)
levels and aortic valve calcification and stenosis, suggesting a causal role for Lp(a) in valvular calcification. However, the relationship between Lp-PLA2 and Lp (a) is interesting. 24
1.2.2 Lp-PLA2 and PAF
When it was first discovered, and before joining the PLA2 family, Lp-PLA2 was initially known as Platelet activating factor acetylhydrolase (PAF-AH). PAF-AH was shown to hydrolyze the ester bond at the sn-2 position of PAF, producing LysoPAF and acetate. PAF-AH was known for its potency in deactivating PAF. Today, it is classified as group VIIA, and is more commonly known as lipoprotein-associated phospholipase A2 (Lp-PLA2) due to its larger substrate specificity including PAF and oxLDL. 17
First described in 1972, platelet-activating factor (PAF) is a phospholipid that is produced by various cell types (platelets, neutrophils, endothelial cells, and others) and acts as a communication mediator that transmits signals between cells. Similar to Lp-PLA2, the name PAF is somewhat misleading because it does not fully describe PAF biological activity. While it activates platelets, PAF has several physiological and pathological effects; it has shown to cause uterine contractions, bronchoconstriction, affect the vascular system (lowering blood pressure, reducing cardiac output, increasing vascular permeability), with a possible effect on the endocrine, gastrointestinal, and other systems. 25, 26 PAF has been linked to the inflammatory process as a proinflammatory molecule that binds to the PAF membrane receptor expressed on various cells, activates multiple signalling pathways (platelets, neutrophils, macrophages) that could trigger an inflammatory response that may include platelet aggregation, apoptosis, and vascular remodelling.
Later, scientists discovered other phospholipids that have a resembling structure to PAF and function in a similar pattern as signalling molecules. The biologic effects of “PAF-like” molecules include platelet activation, PPAR activation, and stimulation of mitosis. Normal cells deactivate PAF and PAF-like molecules by intracellular PAF-AH, while extracellular PAF-AH (Lp-PLA2) also deactivates oxLDL in the arterial wall. 11, 27
Figure 1. Hydrolysis of PAF by Lp-PLA2 (PAF-AH). Chemical structure for PAF was obtained from Lipid maps. 28
1.2.3 Substrate specificity
When it comes to substrates, it seems that Lp-PLA2 has broad specificity. An exact substrate for Lp-PLA2 is not yet well identified despite several studies. Although PAF-AH, by definition, has higher affinity towards PAF, it is now known that Lp-PLA2 /PAF-AH substrates are not limited to PAF, but also include PAF-like molecules which are also phospholipids such as oxidized phospholipids, esterified isoprostanes, PEIPC and PC-hydroperoxides. 15
Figure 2. Hydrolysis of an oxidized phospholipid (POVPC) by Lp-PLA2. Chemical structure for POVPC was obtained from Lipid maps. 28
Min et al reported that diglycerides, triglycerides and acetylated alcohols are also potential
substrates for Lp-PLA2. With an extensive possible structure of an oxLDL molecule, Lp-PLA2 recognizes substrates that have a structure of a glyceride derivative with a hydrophobic chain at the sn-1 position and an ester at the sn-2 position. 29 It has also been suggested that Lp-PLA2 residues 189-239 are responsible for substrate specificity. 21
Despite the large number of potential substrates that could be targeted by Lp-PLA2, it seems that it targets certain phospholipids. Previous work has identified several truncated PC oxidation products as main substrates of Lp-PLA2. 14
The specificity of the enzyme’s reaction can be linked to length of the fatty acyl on the sn2 position of the phospholipid. Action of Lp-PLA2 seems to be more specific to fatty acyl chains of up to nine methylene groups. Phospholipids with longer chains (containing more than nine methylene groups) seem to be exempted from the action of PAF-AH, unless they are oxidized. 14, 30
1.2.4 Products of Lp-PLA2 and their biologic effects
In order to neutralize PAF, Lp-PLA2 cleaves at the sn2 position of PAF to produce lysoPAF, a biologically inactive product, and acetate.
A main product of Lp-PLA2 action on oxidized phospholipids is lysophosphatidylcholine (LPC), in the form of saturated and monounsaturated LPCs. LPC is produced in the plasma by 2 other pathways: LCAT, and another phospholipase A2: sPLA2 which is classified as group IIa. With the production of LPC, a series of proinflammatory consequences escalate the inflammatory process and involves different cells. Those can be described by attracting
monocytes, stimulating arachidonic acid release, upregulating cytokines and adhesion molecules, and induction of apoptosis. 31
With the additional effect of certain enzymes that target LPC, lysophosphatidic acid (LPA) is produced, consequently leading to platelet activation. 11 LPC also acts as an intermediate product that could lead to the reproduction of PAF through the enzyme lysophosphatidylcholine acyltransferase (LPCAT), amplifying the inflammatory process. 27 The other product of Lp-PLA2 action on oxidized phospholipids is free fatty acids (FFA). Those are also have been shown to be proinflammatory as they act as monocyte chemoattractants. 32
1.2.5 Regulation of Lp-PLA2
The PLA2G7 gene located on chromosome 6p21.1-p12 encodes Lp-PLA2. PLA2G7 is expressed in different tissues in the following decreasing order: thyroid, leukocytes, prostate, ovaries, lung, and others. 33, 34 Lp-PLA2 has been associated with the pathophysiology of several diseases and syndromes such asthma, atopy, dementia, coronary artery disease (CAD), heart failure, and stroke. 21
To date, we do not have a complete understanding of Lp-PLA2 regulation. Taking in consideration its broad substrate specificity and controversial function, it seems that the regulation of Lp-PLA2 is multifactorial. Several studies concluded that Lp-PLA2 expression is dependent on cell differentiation and inflammatory mediators. 35, 36 A recent study demonstrated that serum amyloid A (SAA) significantly increases Lp-PLA2 expression in THP-1 cells and ApoE knock out mice. 37 Lp-PLA2 activity in the plasma can also be estrogen regulated. 38
As the main producers of Lp-PLA2, studies targeted Lp-PLA2 expression by macrophages in atherosclerotic plaques as well as the plasma. At the transcriptional level, Ferguson et al showed that Lp-PLA2 mRNA levels were elevated in macrophages (especially M1 macrophages) but not in their precursors, the monocytes, and that expression increases post differentiation. Moreover, the study found Lp-PLA2 mRNA and protein levels to be higher in foam cells as compared to macrophages 36. Another study looked at the regulation of Lp-PLA2 in macrophages and found that interferon gamma (IFN-γ) - a proinflammatory molecule - and lipopolysaccharide repressed Lp-PLA2 synthesis 39.
Previous studies have shown a higher expression of Lp-PLA2 in aortic plaques 40. A recent study by Mahmut et al has shown a several fold increase in mRNA expression for Lp-PLA2 in patients with calcific aortic valve disease (CAVD) as compared to subjects with non-calcified aortic valves. The same study reported a higher plasma Lp-PLA2 activity in CAVD patients as compared to controls, and a higher Lp-PLA2 activity in stenotic valves than in non-mineralized valves 41. We do not know much about its regulation in the plasma, although oxidized phospholipids have been shown to play a role in its regulation through several pathways.
OxLDL appears to be a main player in the regulation of Lp-PLA2 expression. A study by
Wang et al suggested that Lp-PLA2 upregulation was due to the presence of its substrates (oxidized PC species), but not its products. In the same study, the authors noticed that PAF didn’t exert a similar effect on Lp-PLA2 expression 35, while other studies showed that PAF. Finally, it is worthwhile mentioning that several studies have shown that despite Lp-PLA2 being regulated by several pathways, it does not act as an acute phase reactant. 19, 36, 42
1.3 Lp-PLA2 animal models
Because atherosclerosis is a complex process, choosing the suitable animal model depends on the specific aspect under study. 43 The mouse is probably not the best model for Lp-PLA2 and atherosclerosis studies, because their lipid profile is different from humans, and as a result the distribution of Lp-PLA2 is also different. Despite that fact, Lp-PLA2 and a specific Lp-PLA2 inhibitor, Darapladib were studied in LDLR-deficient mice and homozygous ApoE-deficient mice. Both studies showed that Lp-PLA2 inhibition by Darapladib reduces inflammation and is associated with a decrease in plaque formation. 44, 45
A study by Wilensky et al preferred a pig model to study Lp-PLA2 46, where the authors mention that the reasons for choosing the pig model rather than the mouse model were the similarity of plasma lipoprotein profile in pigs to humans, the similarity of Lp-PLA2 distribution in pigs to humans, the better ability to study coronary lesions in those models compared to mice. In addition to those points, pig models had other advantages: anatomical resemblance of their cardiovascular system to humans, the resemblance of their atherosclerotic lesions to human lesions, their bigger size (bigger coronaries, bigger plaques), less need for genetic manipulation, and they can have atherosclerotic lesions even with chow diet (although slower) without the need for induction with high fat diet. In preclinical studies, the use of large animals (pigs, dogs, monkeys) is preferred for testing the efficacy of new agents such as Darapladib. The pig is often selected because of its similarities in terms of metabolism and cardiovascular system to that of man. Thus, the pig model appears a more suitable model for atherosclerotic studies compared to mice or rabbits. 43, 47
1.4 Lp-PLA2 and statins
Statins have shown to reduce both the risk of cardiovascular events (by around 30%) and mortality. Those agents appear to play a role in limiting the atherosclerotic process by decreasing the levels of circulating LDL-cholesterol in the plasma, and possibly through other mechanisms, although not yet confirmed. Several studies show that Lp-PLA2 levels are significantly decreased after taking lipid-lowering medications. 22, 48 It is noticed that patients who were on high doses of statins (HMG-CoA reductase inhibitors) had reduced Lp-PLA2 mass and activity of about 20% to 30%, with Atorvastatin being more effective than other statins. 19, 49, 50 In one of the mentioned studies, Atorvastatin did not affect Lp-PLA2 activity on HDL-C which confirms that statins do not influence Lp-PLA2 production, but rather increased LDL-C clearance. 51 Statins lower Lp-PLA2 levels. The role of the specific Lp-PLA2 inhibitor, Darapladib, has been questioned in clinical trials.
1.5 Controversy of Lp-PLA2 function
The role that Lp-PLA2 plays in CVD is still not very clear. To date, two main opposite roles for the enzyme in atherosclerosis have been suggested: anti-inflammatory and proinflammatory. One view holds that the substrates for Lp-PLA2 action, such as PAF and oxPC are known to be proinflammatory molecules, thereby emphasizing the antiatherogenic properties of Lp-PLA2. However, the by-products of Lp-PLA2 action are also proinflammatory molecules. 52 Several studies have shown high expression of Lp-PLA2 and LPC in unstable plaques. Moreover, they have shown correlation between increased Lp-PLA2 mass and activity and increased risk of CAD, stroke, and death. 40, 53
Overexpression of Lp-PLA2 and its products in atherosclerotic plaques could be to the fact that
PLA2G7 is upregulated by several inflammatory mediators that are abundant in an
inflammatory milieu. Moreover, in vivo studies have shown reduced inflammation with recombinant Lp-PLA2, which supports an anti-inflammatory role 21. A study conducted by Heart Protection Study Collaborative Group concluded that lipoproteins have high influence on the association between Lp-PLA2 and risk of coronary events. With adjustment for apoB levels, the association between the two became trivial 54.
While elevated Lp-PLA2 plasma levels are associated with increased cardiovascular risk, HDL-associated Lp-PLA2 is associated with a lower cardiovascular risk, as observed in 524 stable CAD patients in 3 year follow up study. That supports a more recent interpretation of those observations suggesting that Lp-PLA2 on HDL might participate in the antioxidant function of HDL and has an overall anti-inflammatory role, while Lp-PLA2 on LDL has a proinflammatory function 52.
Taking different points in consideration, it is not clear whether the inhibition of Lp-PLA2 is beneficial in decreasing cardiac risk and mortality, as causality cannot be justified according to those associations. To reconcile this controversy, it is conceivable that the actions of Lp-PLA2 maybe beneficial in one environment, but deleterious in another. Thus, we can postulate that the effects of Lp-PLA2 maybe beneficial in specific tissues e.g. plasma, but maybe deleterious in the plaque, where the molecules resulting from its action may then trigger secondary messengers, and further aggravate inflammation.
1.6 Epidemiologic studies of Lp-PLA2
During the past decades, studies repeatedly explored the association between Lp-PLA2 and risk of CVD. In 2010, the Lp-PLA2 collaboration studies, a meta-analysis, evaluated the records of 79,036 subjects of 32 prospective studies and showed positive correlation between Lp-PLA2 and the risk of CAD, stroke, and death from vascular and nonvascular causes. 55 A more recent study by Gonçalves et al demonstrated significant correlations between Lp-PLA2, LPC species and LPA with several inflammatory mediators (IL-1β, IL-6, MCP-1, macrophage inflammatory protein-1β) and anti-inflammatory mediator (IL-10) in the atherosclerotic plaques in humans. 31 A previous study showed a higher carotid plaque expression of Lp-PLA2 and a higher concentration on LPC in symptomatic patients as compared to non-symptomatic patients undergoing carotid endarterectomy. 40 Vicker’s et al showed that Lp-PLA2 and oxLDL were highly correlated and expressed in areas with greater potential of plaque rupture artery (bifurcation and internal segments of the carotid artery). 56 Two recent studies investigated Lp-PLA2 in the ACS setting and noticed strong positive correlations between Lp-PLA2 and LDL-C, ApoB with no association with hs-CRP and troponins. Meanwhile, studies have shown that the correlation between Lp-PLA2 and HDL-C was either weak or inverse. 19, 40, 57
In the setting of familial hypercholesterolemia (FH), a recent study by Tellis et al that included 53 patients and detected significant elevation in Lp-PLA2 mass and activity. 22 In addition, few case studies reported high plasma Lp-PLA2 activity in patients with Tangier’s Disease (TD), which is manifested by low levels of circulating HDL and defective cholesterol efflux caused by mutations in the ABCA1 gene, leading to higher risk of CVD 58. In a study that included almost 500 subjects, increased Lp-PLA2 levels were reported in heart failure and aortic
aneurysm patients. 59 In 48 patients with severe aortic stenosis without overt atherosclerosis, inverse correlation between Lp-PLA2 and aortic valve areas was reported, in addition to significant elevations in Lp-PLA2 levels. 60
The controversy of Lp-PLA2 function extends to genetic studies. A relatively common
PLA2G7 mutation of (V279), has been identified in several populations, and has shown to be
more prevalent in East Asian populations (especially in Japan) than other Asian or European countries such as Turkey. This mutation results in partial or complete loss of function of Lp-PLA2. Studies evaluating this mutation reported conflicting results, with more studies emphasizing a proinflammatory role for Lp-PLA2 in the carriers of this mutation. 61-63
Finally, Brilakis reported that race and sex influence Lp-PLA2 levels, suggesting taking those factors in consideration when measuring Lp-PLA2 levels. 64 It also seems that diet and lifestyle measures such as smoking, weight and other factors might affect Lp-PLA2 activity. 65 Another recent study by Da Silva et al reported that Lp-PLA2 activity is influenced by glucose levels, HDL size and Apo B/Apo AI ratio in obese adolescents. 66
1.7 Lp-PLA2 inhibition: the past, the present, and the future
At time of discovery, Lp-PLA2 or better known at that time as PAF-AH was thought to have exclusive anti-inflammatory effects. PAF-AH studies showed significant reduction of severe inflammation in vivo and in vitro. It is ironic that what is known today as a proinflammatory enzyme was not very long ago known as an anti-inflammatory enzyme and was suggested as a treatment for inflammation. Studies suggesting a rather anti-inflammatory character of Lp-PLA2 emphasize its role on degrading PAF and oxidized phospholipids on lipoproteins. At the
present time, the epidemiological and experimental data does not allow to solve the controversy on the protective or the proatherogenic role of Lp-PLA2.
An argument can be made that despite the positive correlation between Lp-PLA2 and increased risk of CVD, the information present is not enough to imply causality. 55 The only way to show causality is by undergoing clinical trials.
A normal level of Lp-PLA2 is <200 ng/mL, while a level of ≥223 ng/mL is considered high and current guidelines do not warrant Lp-PLA2 measurement as part of routine testing in patients with low risk for CAD. 67
Phospholipase A2 inhibition presents an appealing therapeutic target. Two experimental phospholipase A2 inhibitors exist: Darapladib (Lp-PLA2 inhibitor) and Varespladib (sPLA2 inhibitor). When we compare the two agents, we notice that they inhibit different PLA2 isoforms. Darapladib uniquely inhibits Lp-PLA2; Varespladib inhibits several secreted PLA2 isoforms (Group IIa, Group V and Group X), which have shown to have proinflammatory effects in atherosclerotic plaque formation. Similar to Lp-PLA2, they produce LPC and NEFA. 18 In addition to Lp-PLA2 inhibition, Darapladib lowered the expression of 24 genes, several of which are inflammatory markers. 46
The two agents (Darapladib, Varespladib) have been evaluated in several clinical trials, and in different in vitro and in vivo studies. Both have shown efficacy in inhibiting the enzymes in vitro, but lacked efficacy in humans. 44, 45, 68 In a phase II randomized clinical trial that sought to look at the effects of Darapladib in patients with stable coronary heart disease or coronary heart disease risk equivalent, positive correlation was present between LDL-C and Lp-PLA2 activity, while a negative correlation between HDL-C and Lp-PLA2 was noticed. 49 In the phase II (Integrated Biomarker And Imaging Study– 2) IBIS-2 trial, more than 300 patients
with angiographically documented coronary heart disease and another risk factor were randomized to darapladib 160 mg/d or placebo for 12 months to examine the effects of Darapladib, where the results showed reduction in plaque areas and necrotic core in the treatment group as compared to the control group.
IBIS-2 paved the road for Darapladib to undergo two large randomized phase III clinical trials. The STABILITY (STabilisation of Atherosclerotic plaque By Initiation of darapLadIbTherapY) trial studied the effects of chronic treatment with Darapladib on the incidence of major adverse cardiovascular events (MACE) in high-risk patients with chronic CHD in 15,828 men or women with chronic coronary heart disease. STABILITY failed to meet the primary end points (relative risk reduction of 6%), despite some reductions in some of the secondary endpoints. 68, 69 The SOLID-TIMI 52 trial (The Stabilization Of pLaques usIng Darapladib-Thrombolysis In Myocardial Infarction 52) evaluated if Darapladib can lower the chances of having a cardiovascular event when treatment is started within 30 days after ACS. This study involved 13,000 men or women hospitalized for ACS. 70 Overall, Darapladib appears to be well tolerated by patients, and has not been reported to have adverse effects, except for minor effects like diarrhea and odour. 71 Nevertheless, SOLID-TIMI 52 suffered the same fate as STABILITY, and failed to meet its primary end point. 72
The other agent, Varespladib, did not have fruitful results. The phase III trial (Vista-16), which evaluated the safety and efficacy of Varespladib treatment on morbidity and mortality when added to Atorvastatin in 5,189 subjects with ACS, was unexpectedly terminated in March 2012. Varespladib results were consistent in all the previous trials it underwent, but it stumbled in Vista-16 due to unknown reasons at present. 73, 74
Lp-PLA2 signifies a relation between oxidized lipoproteins (oxLDL) and inflammation and plaque susceptibility to rupture. It is highly possible that Lp-PLA2 is a physiological responder rather than a cause of inflammation. Will Lp-PLA2 have the same fate as hsCRP? The new results of the randomized clinical trials urge us to believe that Darapladib and other Lp-PLA2 inhibitors may not see the light at the end of the tunnel. Until proven otherwise, Lp-PLA2 is of limited benefit in a clinical setting.
Chapter II: Clinical Research
Acute coronary syndromes are underlined by a long-term inflammatory state. Lp-PLA2 functions by oxidizing modified phospholipids on the surface of the lipoprotein, an action that results in producing and accumulation of molecules (namely lysophosphatidylcholine and non-esterified fatty acids) within the vessel wall, contributing to unstable plaque formation and aggravating inflammation. Multiple studies have observed an association between the role of Lp-PLA2 in plaque formation and increased risk of coronary artery disease, suggesting an inflammatory role for Lp-PLA2.
The hypothesis was that the inflammatory milieu accompanying Acute Coronary Syndromes alters PLA2 levels and distribution (HDL and LDL). In the present study, we examined Lp-PLA2 mass in patients with ACS acutely (within 48 hours) and again at ACS recovery (12 weeks after their first presentation). Stable CAD and healthy control subjects were used for comparison.
In the plasma, Lp-PLA2 is mainly distributed between LDL-C and HDL-C, with a greater percentage on LDL-C. A secondary objective was to compare the distribution of Lp-PLA2 in the plasma and among lipid fractions (HDL and LDL) in ACS acute and ACS recovery groups. To answer our research questions, we used two different separation techniques including ultracentrifugation (UTC) and high-performance liquid chromatography (HPLC).
Chapter III: Clinical Research
Lipoprotein-Associated Phospholipase A
2(Lp-PLA
2) in Acute
Coronary Syndrome: Relationship with low-density lipoprotein
cholesterol
Bashar Jabor, Hong Choi, Isabelle Ruel, Anouar Hafiane, Walid Mourad,
and Jacques Genest
Can J Cardiol. 2013 Dec;29(12):1679-86.doi:10.1016/j.cjca.2013.09.026.
Epub 2013 Oct 2
Abstract
Background: Lipoprotein-associated phospholipase A2 (Lp-PLA2) might play a role in the formation of vulnerable atherosclerotic plaques. Its plasma distribution and mass in subjects with acute coronary syndrome (ACS) has yet to be characterized.
Methods: We compared plasma levels of Lp-PLA2 in 24 patients within 48 hours of an ACS (acute) and 12 weeks after (recovery), in 26 patients with stable coronary artery disease and in 10 normal healthy control subjects. Lp-PLA2 mass was determined using enzyme-linked immunosorbent assay.
Results: The ACS patients (mean age 57 ± 8.7 years) had high-sensitivity C-reactive protein
(hsCRP) levels of 30.46 ± 57.57 mg/L (ACS acute) vs. 1.69 ± 1.32 mg/L (ACS recovery). Plasma Lp-PLA2 levels were significantly higher in ACS acute subjects than in ACS recovery subjects (143.13 ± 60.88 ng/mL vs. 88.74 ± 39.12 ng/mL; P < 0.0001). Interestingly, stable coronary artery disease patients had higher Lp-PLA2 levels than ACS recovery patients (121.72 ± 31.11 ng/ mL vs. 88.74 ± 39.12 ng/mL; P = 0.0018). There was a strong correlation between Lp-PLA2 and low-density lipoprotein (LDL) cholesterol (LDL-C) (r = 0.709; P < 0.0001) or changes in LDL (r = 0.449; P = 0.027), suggesting that the major determinant of plasma Lp-PLA2 is LDL-C. No significant correlations were observed between Lp-PLA2 and hsCRP or density lipoprotein (HDL) cholesterol. When separated using high-performance liquid chromatography, > 65%-70% of Lp-PLA2 mass was within the apolipoprotein B-containing lipoprotein fraction, with approximately 30%-35% on HDL fraction, with no significant change in distribution between ACS acute and recovery.
Conclusions: Subjects with an ACS have markedly increased Lp-PLA2 levels acutely related to LDL-C levels.
Résumé
Introduction: La phospholipase A2 associée aux lipoprotéines (Lp-PLA2) pourrait jouer un rôle dans la formation de plaques athéroscléreuses vulnérables. Sa distribution plasmatique et sa masse chez les sujets ayant un syndrome coronarien aigu (SCA) a encore à être déterminée.
Méthodes: Nous avons comparé les concentrations plasmatiques de Lp-PLA2 chez 24 patients dans les 48 heures d’un SCA (aigu) et dans les 12 semaines après (rétablissement), chez 26 patients ayant une coronaropathie stable et chez 10 témoins en bonne santé. La masse de la Lp-PLA2 a été déterminée à l’aide de l’immunoabsorption par enzyme liée.
Résultats: Les patients ayant un SCA (âge moyen de 57 ± 8,7 ans) ont eu des concentrations
de protéine C réactive élevées de 30,46 ± 57,57 mg/l (SCA aigu) vs 1,69 ± 1,32 mg/l (rétablissement du SCA). Les concentrations plasmatiques de la Lp-PLA2 ont été significativement plus élevées chez les sujets ayant un SCA aigu que chez les sujets rétablis d’un SCA (143,13 ± 60,88 ng/ml vs 88,74 ± 39,12 ng/ ml; P < 0,0001). Fait intéressant, les patients ayant une coronaropathie stable ont eu des concentrations de la Lp-PLA2 plus élevées que les patients rétablis d’un SCA (121,72 ± 31,11 ng/ml vs 88,74 ± 39,12 ng/ml; P = 0,0018). Il existait une forte corrélation entre la Lp-PLA2 et le cholestérol à lipoprotéines de faible densité (C-LDL; r = 0,709; P < 0,0001) ou les changements dans le LDL (r = 0,449; P = 0,027), ce qui suggère que le déterminant majeur de la Lp-PLA2 plasmatique est le C-LDL. Aucune corrélation significative n’a été observée entre la Lp-PLA2 et la protéine C réactive hautement sensible ou le cholestérol à lipoprotéines de haute densité (HDL). À la séparation par la chromatographie à haute performance, > 65%-70% de la masse de la Lp-PLA2 a été dans la fraction de la lipoprotéine contenant l’apolipoprotéine B, dont approximativement 30%
-35% sur la fraction de HDL, et aucun changement significatif dans la distribution entre le SCA aigu et le rétablissement du SCA.
Conclusions: Les sujets ayant un SCA ont en phase aigue une augmentation significative des
Lipoprotein-associated phospholipase A2 (Lp-PLA2) is a novel biomarker that is postulated to play a role in the formation of atherosclerotic plaques and might lead to plaque instability. Human Lp-PLA2 is encoded by the PLA2G7 gene. 75 It is produced by many cells, inflammatory (monocytes/macrophages, mast cells) and noninflammatory cells (liver Kupffer cells, platelets, erythrocytes), but it appears that macrophages are the main producers. 23 Similar to other phospholipase A2 enzymes, Lp-PLA2 catalyzes the hydrolysis of the ester bond at the sn-2 position of phospholipids. Lp-PLA2 is bound to lipoproteins, predominantly to low-density lipoprotein (LDL) (approximately 80%), with a significantly smaller portion on high-density lipoprotein (HDL) (approximately 20%), 76 hence the name, Lipoprotein-associated phospholipase A2. Originally described as platelet activating factor acetyl hydrolase, Lp-PLA2 cleaves the oxidized glycerophospholipid acetyl-glyceryl-ether-phosphorylcholine in the sn-2 position to yield lysophosphatidylcholine (LPC) and an acetyl group. However, the substrate specificity of Lp-PLA2 is not well defined and it is thought to inactivate highly reactive oxidized phospholipids on lipoproteins. The products of the Lp-PLA2 action, LPC and nonesterified fatty acids, are also known to be proinflammatory and to be involved in cell signalling.31Lp-PLA2 thus represents a link between oxidized lipoproteins, inflammation, and plaque instability. In the arterial wall, LDL is modified into oxidized lipoproteins under oxidative stress, modifying some of the phospholipids on the LDL particle. The production of oxidized phospholipids activates LDL-bound Lp-PLA2. Lp-PLA2 hydrolyzes those oxidized phospholipids (mainly oxidized phosphatidylcholine) and generates 2 different molecules: LPC and nonesterified fatty acids. 23
Lp-PLA2 is found in atherosclerotic plaques using immunohistology and its presence correlates with inflammatory cells and histological characteristics of plaque instability.77 A
meta-analysis of Lp-PLA2 in cardiovascular disease (CVD) reveals that Lp-PLA2 levels (mass and activity) are significantly associated with each other and with proatherogenic lipid markers. Lp-PLA2 levels are significantly related to CVD risk in a continuous and graded manner. 55 Because Lp-PLA2 levels predict future cardiovascular events, the measurement of Lp-PLA2 might be considered (Class IIb) in intermediate-risk asymptomatic adults. 78
The effects of lipid-lowering therapies in Lp-PLA2 levels have been previously examined, 48, 79 and the data show that statins significantly lower Lp-PLA2 levels. Albert et al showed that approximately 6% of the variability in the response of Lp-PLA2 to pravastatin was accounted for by a reduction in LDL cholesterol (LDL-C) levels. 80 In most reports, the effect of statins on Lp-PLA2 levels was significantly attenuated after adjusting for changes in LDL-C levels, reflecting the strong affiliation between these 2 substances in plasma. A recent study of Lp-PLA2 levels in the first 3 days after an acute coronary syndrome (ACS) reveals that Lp-Lp-PLA2 levels probably increase acutely, but that the subsequent change in levels correlate with the change in LDL-C. 57 Previous data have not shown any significant correlations between reductions in Lp-PLA2 levels and changes in HDL cholesterol (HDL-C) concentrations.
The purpose of the present study was to examine plasma levels of Lp-PLA2 in ACS and its relationship to high-sensitivity reactive protein (hsCRP) and lipoprotein levels. Unlike C-reactive protein, Lp-PLA2 does not seem to behave as an acute-phase reactant. 42 We hypothesized that the inflammatory milieu that is associated with an ACS might alter plasma levels of Lp-PLA2. The main objective of this study was to determine plasma levels and distribution of Lp-PLA2 at the time of an ACS (within 48 hours of onset of symptoms) and again, 12 weeks later, compared with patients with stable, chronic coronary artery disease
(CAD) (> 1 year after the event). We also determined the concentration of Lp-PLA2 in lipoprotein fractions.
Methods
Study subjects.
Patients, men and women aged between 18 and 80 years, were selected from the McGill University Health Center, Canada. Subjects within the ACS acute group were sampled within 48 hours of symptom onset (n = 24) according to current diagnostic criteria. 81 The diagnosis of ACS was confirmed in a clinical assessment by a cardiologist (J.G.), including electrocardiographic changes (non-ST and ST segment elevation acute myocardial infarction), troponin I elevation, and the presence of CAD on coronary angiography. The same patients were resampled 12 weeks later (ACS recovery; n = 24). The 12-week time point was selected because most of the metabolic, lipoprotein, and inflammatory abnormalities have resolved by then. 82 As comparison groups, we also examined subjects with CAD and normal healthy control subjects (normals). The CAD group consisted of patients with chronic, stable CAD who had been followed in the lipid clinic for at least 11 year after a cardiovascular event, and the normals consisted of age-matched healthy subjects without CAD. These subjects had no CAD, documented using coronary angiography; all were awaiting elective aortic or mitral valve replacement or repair, and were not taking statin therapy. All subjects with stable CAD continued taking statins at the time of sampling, according to current guidelines and ethics requirements. 83-86 Exclusion criteria in the ACS patients included refusal to participate, inability to return for a 12-week follow-up visit, hemodynamic instability requiring vasopressor support, cardiac support devices, the need for urgent coronary artery bypass surgery, and the lack of documented atherosclerotic CAD, uncontrolled hypertension, triglycerides ≥ 5 mmol/L, severe obesity (body mass index ≥ 35), alcohol intake > 21 drinks per week, and presence of thyroid, hepatic, or renal disease. Subjects were excluded if they
had autoimmune disease or any chronic or acute infectious or inflammatory illness. The research protocol was reviewed and approved by the Research Ethics Board of the McGill University Health Center; all subjects gave written informed consent to participate.
Plasma lipids, lipoprotein lipids, lipoprotein isolation, apolipoprotein B, hsCRP, and Lp-PLA2 mass assays.
Blood was collected from study subjects and control subjects using venipuncture; plasma was separated (2000g, 4 °C for 20 minutes) and multiple aliquots were frozen at -80 °C in 0.5-mL aliquots. Total cholesterol, triglycerides, and HDL-C were determined enzymatically and LDL-C was determined using the Friedewald formula. A sample of plasma (9 mL) was immediately processed for isolation of lipoproteins using ultracentrifugation (UTC). We isolated apolipoprotein B (apoB)-containing lipoproteins (LpB) in the < 1.09 g/mL density fraction and HDL using sequential UTC at a density between 1.09 g/mL and 1.21 g/mL using KBr as a salt. Isolated LpB and HDL fractions were then extensively dialyzed against phosphate-buffered saline, and total protein concentrations in these fractions were measured using a modified Bradford procedure (BioRad, Mississauga, Canada). Apolipoprotein A-1, apoB, and hsCRP concentrations were deter- mined using nephelometry (Behring Nephelometer 100 Analyzer). Lp-PLA2 concentrations (mass) were measured using a commercial Lp-PLA2 enzyme-linked immunosorbent assay (Quantikine ELISA, R&D Systems).
Pools of plasma (125 uL) from the ACS acute and the ACS recovery group (n = 24) were separated into lipoprotein fractions using high-performance liquid chromatography (HPLC) with a Superose 6 10/300 GL column (GE Healthcare). A 150-mM NaCL mobile phase with a
flow rate of 0.4 mL/mL was used for the separation of the sample into seventy-two 400-uL HPLC fractions that were collected. Total cholesterol was determined enzymatically using the Infinity Cholesterol Liquid Stable Reagent (Thermo Electron Corporation) following the manufacturer’s instructions.
Statistical analyses.
Discrete variables are described as mean ± SD. For comparisons between ACS acute and ACS recovery, 2-tailed paired t tests with equal variance were selected. A P value of < 0.05 was considered significant. Pairwise comparisons between ACS acute, CAD, and Normals were considered significant at the 0.05 level. Pearson correlation coefficients were used to examine the relationship between Lp-PLA2 and LDL-C, apoB, hsCRP, HDL, and troponins. Linear regression was used to determine the effect of LDL-C-lowering on Lp-PLA2 levels. All statistical analyses were performed using the GraphPad Prism software V5.0 (La Jolla, CA).
Results
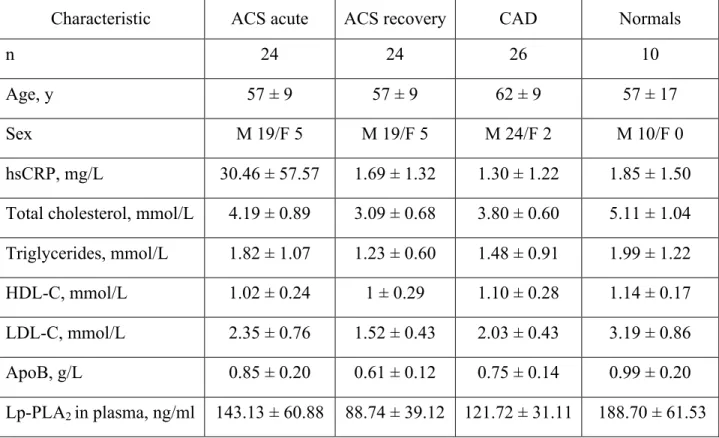
Patient characteristics are summarized in Table 1. There were 24 patients in the ACS groups (19 men, 5 women, mean age 57 ± 9 years), 26 with chronic, stable CAD (24 men, 2 women, mean age 62 ± 9 years) and 10 Normals (10 men aged 57 ± 17 years). Mean hsCRP level in ACS acute subjects was 30.46 ± 57.57 mg/L and decreased to 1.69 ± 1.32 mg/L in ACS recovery, not significantly different from CAD (1.30 ± 1.22 mg/L) and Normals (1.85 ± 1.50 mg/L). Peak troponin I level was 24.54 ± 36.29 ug/L in ACS acute (reference range, < 0.06 mg/L), indicating acute myocardial injury. Plasma levels of LDL-C differed markedly between groups. Approximately half of the patients in ACS acute were taking various doses of statins at the time of sampling; the LDL-C in ACS acute was 2.35 ± 0.76 mmol/L. As per clinical practice guidelines, 83-86 ACS patients were put on a high-dose statin regimen (predominantly atorvastatin 80 mg or rosuvastatin 40 mg) and the LDL-C decreased to 1.52 ± 0.43 mmol/L at 12 weeks (P < 0.0001 ACS acute vs. ACS recovery) (Table 1). CAD patients had a mean LDL-C of 2.03 ± 0.43 mmol/L. Normals were not using statins by design and the mean LDL-C was 3.19 ± 0.86 mmol/L. HDL-C levels were similar between ACS acute, ACS recovery, CAD, and Normals (Fig. 1).
Table 1. Study subject characteristics
Characteristic ACS acute ACS recovery CAD Normals
n 24 24 26 10
Age, y 57 ± 9 57 ± 9 62 ± 9 57 ± 17
Sex M 19/F 5 M 19/F 5 M 24/F 2 M 10/F 0
hsCRP, mg/L 30.46 ± 57.57 1.69 ± 1.32 1.30 ± 1.22 1.85 ± 1.50 Total cholesterol, mmol/L 4.19 ± 0.89 3.09 ± 0.68 3.80 ± 0.60 5.11 ± 1.04 Triglycerides, mmol/L 1.82 ± 1.07 1.23 ± 0.60 1.48 ± 0.91 1.99 ± 1.22 HDL-C, mmol/L 1.02 ± 0.24 1 ± 0.29 1.10 ± 0.28 1.14 ± 0.17 LDL-C, mmol/L 2.35 ± 0.76 1.52 ± 0.43 2.03 ± 0.43 3.19 ± 0.86
ApoB, g/L 0.85 ± 0.20 0.61 ± 0.12 0.75 ± 0.14 0.99 ± 0.20
Lp-PLA2 in plasma, ng/ml 143.13 ± 60.88 88.74 ± 39.12 121.72 ± 31.11 188.70 ± 61.53
Relevant P values are shown in Figures 1 and 2.
ACS, acute coronary syndrome; ApoB, apolipoprotein B; CAD, coronary artery disease; F, female; HDL-C, high-density lipoprotein cholesterol; hsCRP, high- sensitivity C-reactive protein; LDL-C, low-density lipoprotein cholesterol; Lp-PLA2, Lipoprotein-associated phospholipase A2; M, male.
Figure 1. hsCRP and HDL-C (inset) levels in study subjects. Patients with an ACS (ACS
acute) were sampled within 48 hours of presentation. The same patients were sampled 12 weeks later (ACS recovery). Note the increase in hsCRP in the ACS acute group, compared with the recovery, CAD, and normal subjects. ACS, acute coronary syndrome; CAD, coronary artery disease; HDL-C, high-density lipoprotein cholesterol; hsCRP, high-sensitivity C-reactive protein.
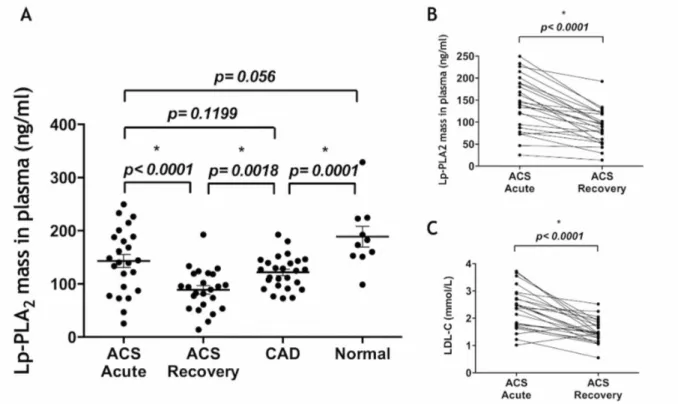
Figure 2. (A) Lp-PLA2 levels in the plasma of study subjects: ACS acute (n = 24), ACS recovery (n = 24), CAD (n = 26), and normal healthy subjects (n = 10). (B) Difference in Lp-PLA2 mass between ACS acute and recovery. (C) Difference in LDL-C between the same groups. ACS, acute coronary syndrome; CAD, coronary artery disease; LDL-C, low-density lipoprotein cholesterol; Lp-PLA2, lipoprotein-associated phospholipase A2.
Plasma Lp-PLA2 levels were significantly increased in ACS acute (143.13 ± 60.88 ng/mL) compared with ACS recovery (88.74 ± 39.12 ng/mL; Fig. 2). CAD subjects had slightly higher levels than ACS recsovery (121.72 ± 31.11 ng/mL) and Normals had Lp-PLA2 levels in the normal range (188.70 ± 61.53 ng/mL), and not significantly higher than the ACS acute subjects (reference range, < 200 ng/mL). 67 In all patients, Lp-PLA2 levels decreased at 12 weeks (Fig. 2B), and paralleled the decrease in LDL-C (Fig. 2C).
UTC was used to separate LpB at a density of 1.006 g/L < LpB <1.09 g/L and HDL d = 1.09 g/L < HDL < 1.21g/L. Only approximately 50% of Lp-PLA2 mass was recovered in the LpB fraction (Supplementary Figure 1A) (43% of total) and 3% in the HDL fraction (data not shown), suggesting that Lp-PLA2 is only loosely bound to lipoproteins and can be sheared off during UTC.
We then separated lipoproteins using HPLC as shown in Figure 3A. The Lp-PLA2 distribution closely matched that of the LDL-C peak. A representative control HPLC analysis from a patient with high triglycerides (Fig. 3B) and high HDL-C (Fig. 3C) confirmed that Lp-PLA2 resides predominantly within LDL. HPLC analysis of serum did not reveal a shift in the distribution of Lp-PLA2 between HDL and LDL. UTC, as previously stated, caused a significant loss of LDL- and HDL- associated Lp-PLA2, making quantification unreliable.
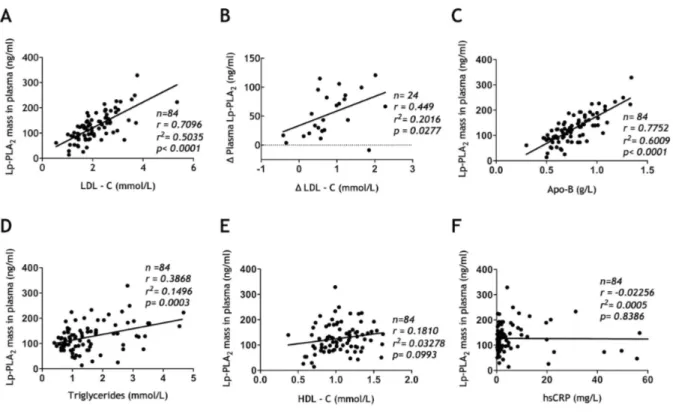
When examining the correlations of Lp-PLA2 in all subjects, there was a strong and highly significant correlation between LDL-C and Lp-PLA2 (r = 0.709; r2 = 0.503; P < 0.0001), suggesting that half the variance in Lp-PLA2 can be explained by LDL-C levels (Fig. 4A and B). Similar data were obtained for total plasma apoB (r = 0.775; r2 = 0.6009; P < 0.0001). The change in Lp-PLA2 also correlated strongly with change in LDL-C between ACS acute and ACS recovery patients (n = 24; r = 0.449; P = 0.027) (Fig. 4B). These data suggest that LDL is
the major determinant of Lp-PLA2. In the 4 groups combined, the correlation between triglycerides and plasma Lp-PLA2 levels was r = 0.3868 (n = 84; P = 0.0003) (Fig. 4D). No significant correlation was observed between HDL-C and Lp-PLA2 (r = 0.181; P = 0.099) (Fig. 4E), between hsCRP and Lp-PLA2 (r = -0.023; P = 0.839) (Fig. 4F) or between troponin I and Lp-PLA2 in ACS acute (data not shown). When we examined groups separately, there was no correlation between hsCRP and Lp-PLA2 levels. The correlations for all variables are shown in Table 2.
Figure 3. Distribution of Lp-PLA2 mass. (A) Lp-PLA2 and cholesterol mass assays performed after HPLC separation of pooled plasma samples from ACS acute (n = 24) and ACS recovery (n = 24). Note that the distribution of Lp-PLA2 parallels that of LDL predominantly. (B) and
(C) show the distribution of Lp-PLA2 in 2 control subjects, a normo-lipidemic subject (B) and 1 with elevated triglyceride levels (C), showing that Lp-PLA2 remains predominantly within the LDL-size fraction. ACS, acute coronary syndrome; HPLC, high-performance liquid chromatography; LDL, low-density lipoprotein; Lp-PLA2, lipoprotein-associated phospholipase A2; OD, optical density.
Figure 4. Correlations between Lp-PLA2 and LDL-C (A); change in LDL-C (B); Apo-B (C); HDL-C (D); triglycerides (E), and hsCRP (F). Apo-B, apolipoprotein B; HDL-C, high-density lipoprotein cholesterol; hsCRP, high-sensitivity C-reactive protein; LDL-C, low-density lipoprotein cholesterol; Lp-PLA2, lipoprotein-associated phospholipase A2.
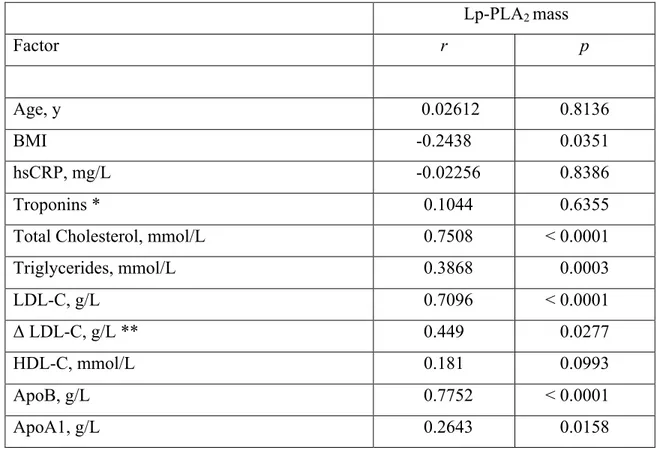
Table 2. Pearson correlation coefficients of Lp-PLA2 mass with cardiovascular risk factors in the study subjects.
Lp-PLA2 mass Factor r p Age, y 0.02612 0.8136 BMI -0.2438 0.0351 hsCRP, mg/L -0.02256 0.8386 Troponins * 0.1044 0.6355
Total Cholesterol, mmol/L 0.7508 < 0.0001
Triglycerides, mmol/L 0.3868 0.0003 LDL-C, g/L 0.7096 < 0.0001 Δ LDL-C, g/L ** 0.449 0.0277 HDL-C, mmol/L 0.181 0.0993 ApoB, g/L 0.7752 < 0.0001 ApoA1, g/L 0.2643 0.0158
ACS, acute coronary syndrome; ApoA1, apolipoprotein A1; ApoB, apolipoprotein B; BMI, body mass index; HDL-C, high-density lipoprotein cholesterol; hsCRP, high-sensitivity C-reactive protein; LDL-C, low-density lipoprotein cholesterol; Lp-PLA2, lipoprotein-associated phospholipase A2.
* For ACS acute only (n = 24).
Discussion
In the present study, we showed that Lp-PLA2 in human is not an acute phase reactant and does not increase in acute inflammation. We also showed that Lp-PLA2 mass is closely related to LDL and LpB, with only a minor fraction within HDL. This has important implications if Lp-PLA2 is considered a therapeutic target. Lowering LDL-C with statins has a marked and proportional effect on Lp-PLA2 mass.
A meta-analysis of the association between Lp-PLA2 mass or activity in plasma or serum shows that Lp-PLA2 levels predict recurrent cardiovascular risk. A small molecule, darapladib, is a specific inhibitor of Lp-PLA2 function and is currently undergoing clinical trials in patients with CAD. In the Stabilization of Atherosclerotic Plaque by Initiation of Darapladib
Therapy (STABILITY) trial, more than 15,000 patients with established CAD and another risk
factor were randomized to darapladib 160 mg/d or placebo and a back- ground of appropriate lipid-lowering therapy with statins. 68 In the Stabilization of Plaques using Darapladib -
Thrombolysis in Myocardial Infarction 52 (SOLID-TIMI 52) trial, 13,000 patients with an
ACS have been randomized to darapladib or placebo. 70 Both trials are expected to be complete in 2013 and 2014 respectively. Importantly, both trials will be performed in patients treated to strict LDL-C goals. Considering the redundancy of phospholipases A2 - there are 15 known phospholipase A2 groups comprising as many isoforms in humans 87; it is not certain that this approach will prevent CVD.
A similar approach was used with varespladib, an inhibitor of the secretory phospholipase A2 isoforms IIa, V, and X. In the Vascular Inflammation Suppression to Treat Acute Coronary Syndrome-16 Weeks (VISTA-16) trial, varespladib failed to meet its primary end point of CVD reduction. 70, 73, 74, 88
Previous groups have examined Lp-PLA2 levels in the early phase of an ACS. Dullaart et al reported that Lp-PLA2 levels in ACS did not differ from those found in patients with noncardiac chest pains. Interestingly, Lp-PLA2 levels were greater in a subset of patients with ST segment elevation myocardial infarction. The study did not control for baseline lipid values. 89 Ostadal et al recently reported that Lp-PLA2 levels were increased in the initial hours of an ACS, compared with in the following days. They reported an observation similar to ours, showing a relationship between LDL-C and Lp-PLA2 and a lack of correlation with C-reactive protein and troponin I. 57
The distribution of Lp-PLA2 on lipoproteins was demonstrated many years ago, 76, 90 and revealed that a minor percentage (approximately 20%) was associated with HDL. In a prospective study, Lp-PLA2 in plasma and within HDL was determined in more than 500 patients who were followed for 3 years. Although Lp-PLA2 in plasma levels were associated with worse outcomes, HDL-associated Lp-PLA2 mass was associated with a lower risk (hazard ratio, 0.972; 95% confidence interval, 0.952-0.993; P = 0.010). An even more impressive effect of HDL-associated Lp-PLA2 activity on cardiac death (hazard ratio, 0.689; 95% confidence interval, 0.496-0.957; P = 0.026) was observed. 52
The biology of Lp-PLA2 is only partly understood. Initially described as a platelet-activating factor (or 1-O-alkyl-2-acetyl-sn- glyceryl-3-phosphorylcholine) acetylhydrolase, its physiological role remains uncertain. It is possible that on LDL, Lp-PLA2 hydrolyzes oxidized phospholipids, generating LPC and oxidized lipids that are deleterious within an atherosclerotic plaque, whereas when present on HDL, it neutralizes platelet-activating factor and prevents platelet activation. Lp-PLA2 is present in atherosclerotic plaques, as shown using immunohistochemistry and its presence correlates with histological features of plaque
instability. 77 However, a causal relationship between Lp-PLA2 presence and plaque rupture has not been yet demonstrated. Thus, for the present time, the measurement of Lp-PLA2 mass should be considered a biomarker of atherosclerosis. Its pathological role and potential as a therapeutic target must be further studied.
The main conclusion of the present study is the very close correlation between LDL-C and Lp-PLA2 levels, suggesting that statin therapy might contribute to the reduction in cardiovascular events in a pleiotropic fashion. Our study has several limitations. The sample size was relatively small. We were careful to compare the same patients in ACS acute and ACS recovery groups and to match CAD patients and Normals for age and HDL-C levels. We did not determine Lp-PLA2 activity; however, previous studies have shown a strong correlation between mass and activity. 55, 90Importantly, the quantification of Lp-PLA2 within lipoprotein fractions is limited by the technique used. We have shown that UTC causes loss of Lp-PLA2 mass, most likely because of the shear stress engendered. More problematic is the quantification of Lp-PLA2 within the HDL range. It is extremely difficult to separate HDL particles from plasma proteins using HPLC. Thus, “lipoprotein-free” Lp-PLA2 (i.e. not bound to lipoproteins is very difficult to quantify). In our previous study of HDL proteomics in ACS, Lp-PLA2 was not identified as 1 of the 70 proteins within HDL. 91
references:
23. Tselepis AD, Dentan C, Karabina SA, Chapman MJ, Ninio E. Paf-degrading acetylhydrolase is preferentially associated with dense ldl and vhdl-1 in human plasma. Catalytic characteristics and relation to the monocyte-derived enzyme.
Arteriosclerosis, thrombosis, and vascular biology. 1995;15:1764-1773
31. Goncalves I, Edsfeldt A, Ko NY, Grufman H, Berg K, Bjorkbacka H, Nitulescu M, Persson A, Nilsson M, Prehn C, Adamski J, Nilsson J. Evidence supporting a key role of lp-pla2-generated lysophosphatidylcholine in human atherosclerotic plaque inflammation. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:1505-1512 42. McConnell JP, Hoefner DM. Lipoprotein-associated phospholipase a2. Clinics in
laboratory medicine. 2006;26:679-697, vii
48. Lee SH, Kang SM, Park S, Jang Y, Chung N, Choi D. The effects of statin monotherapy and low-dose statin/ezetimibe on lipoprotein-associated phospholipase a(2). Clinical cardiology. 2011;34:108-112
52. Rallidis LS, Tellis CC, Lekakis J, Rizos I, Varounis C, Charalampopoulos A, Zolindaki M, Dagres N, Anastasiou-Nana M, Tselepis AD. Lipoprotein-associated phospholipase a(2) bound on high-density lipoprotein is associated with lower risk for cardiac death in stable coronary artery disease patients: A 3-year follow-up. Journal of
the American College of Cardiology. 2012;60:2053-2060
55. Lp PLASC, Thompson A, Gao P, Orfei L, Watson S, Di Angelantonio E, Kaptoge S, Ballantyne C, Cannon CP, Criqui M, Cushman M, Hofman A, Packard C, Thompson SG, Collins R, Danesh J. Lipoprotein-associated phospholipase a(2) and risk of coronary disease, stroke, and mortality: Collaborative analysis of 32 prospective studies. Lancet. 2010;375:1536-1544
57. Ostadal P, Vondrakova D, Kruger A, Janotka M, Psotova H, Prucha M. Alteration in lipoprotein-associated phospholipase a2 levels during acute coronary syndrome and its relationship to standard biomarkers. Lipids in health and disease. 2012;11:153
67. Jellinger PS, Smith DA, Mehta AE, Ganda O, Handelsman Y, Rodbard HW, Shepherd MD, Seibel JA, Dyslipidemia ATFfMo, Prevention of A. American association of clinical endocrinologists' guidelines for management of dyslipidemia and prevention of atherosclerosis. Endocrine practice : official journal of the American College of
Endocrinology and the American Association of Clinical Endocrinologists. 2012;18
Suppl 1:1-78
68. The stabilization of atherosclerotic plaque by initiation of darapladib therapy trial (stability).
70. The stabilization of plaques using darapladib-thrombolysis in myocardial infarction 52 trial (solid-timi 52).
73. Vista-16 trial: Evaluation of safety and efficacy of short-term a-002 treatment in subjects with acute coronary syndrome.