Développement d'un alphabet structural intégrant la flexibilité des structures protéiques
Texte intégral
Figure
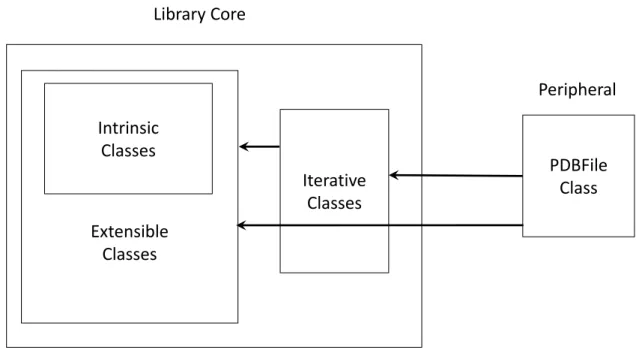
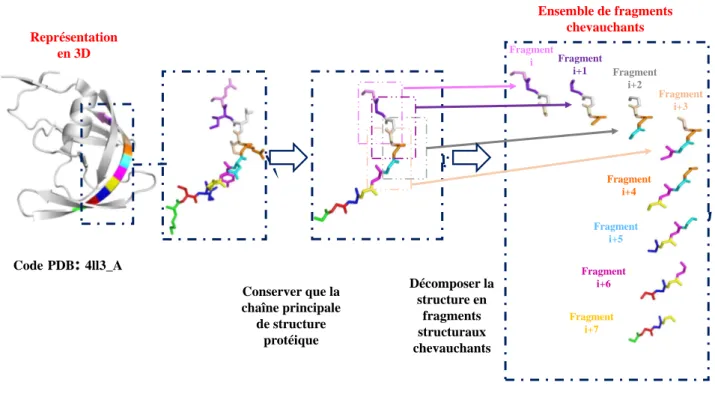
![Figure 3.1 – Géométrie des 27 lettres structurales de l’alphabet structural HMM-SA27 (figure tirée de [142]).](https://thumb-eu.123doks.com/thumbv2/123doknet/2323757.29765/55.892.188.773.654.1043/figure-geometrie-lettres-structurales-alphabet-structural-figure-tiree.webp)

Documents relatifs
La seconde contribution de cette thèse est de proposer des méthodes de calcul com- patibles avec un environnement logiciel et matériel temps-réel permettant de trouver une
En effet, nous allons proposer une fusion de l’approche d’Haralick qui semble plus appropriée, avec celle des lois puissance Zipf et Zipf inverse pour la
Ce module vous offre la possibilité de réaliser une surveillance externe des défauts à la terre, à l’aide d’un transformateur de courant sommateur, plus précise que celle
L‟objectif fondamental de la thèse était de contribuer à une meilleure compréhension des règles qui régissent l‟assemblage des communautés herbacées
Dans le but de proposer une nouvelle approche plus rationnelle du dimensionnement à la ruine des ouvrages de génie civil, Des outils numériques permettant de mettre en œuvre la
La présentation de l 'histoire des sciences pourrait favoriser cette appropriation en permettant de faire un parallèle entre l'évolution des conceptions chez
Plus spécifiquement, l' objectif de ma recherche était de déterminer si des protéines de la voie de signalisation PI3K/ AktINF-KB sont directement liées aux protéines
Cette thèse porte essentiellement sur une étude de la propagation des ondes acoustiques dans un milieu à double porosité, l'objectif étant de proposer une technique non
