HAL Id: tel-03167627
https://tel.archives-ouvertes.fr/tel-03167627
Submitted on 12 Mar 2021HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Caractérisation moléculaire de mécanismes
d’échappement à la sénescence : définition d’une
hétérogénéité de la sénescence
Jordan Guillon
To cite this version:
Jordan Guillon. Caractérisation moléculaire de mécanismes d’échappement à la sénescence : définition d’une hétérogénéité de la sénescence. Médecine humaine et pathologie. Université d’Angers, 2020. Français. �NNT : 2020ANGE0005�. �tel-03167627�
L’auteur du présent document vous autorise à le partager, reproduire, distribuer et communiquer selon les conditions suivantes :
− Vous devez le citer en l’attribuant de la manière indiquée par l’auteur (mais pas d’une manière qui suggérerait qu’il approuve votre utilisation de l’œuvre).
− Vous n’avez pas le droit d’utiliser ce document à des fins commerciales. − Vous n’avez pas le droit de le modifier, de le transformer ou de l’adapter.
Consulter la licence creative commons complète en français : http://creativecommons.org/licences/by-nc-nd/2.0/fr/
Remerciements
Je remercie le Pr. Corinne Abbadie et le Dr. David Bernard d’avoir accepté, en tant que rapporteurs, de consacrer de leur temps à évaluer ce travail et de juger la qualité de mon manuscrit.
Je remercie de même le Dr. Éric Chevet et le Dr. Fabien Gautier d’avoir accepté de faire le déplacement pour faire partie de mon jury de thèse et d’examiner mon travail.
Je tiens à remercier les membres du Rotary Club d’Angers qui par leur don m’ont permis de poursuivre de plusieurs mois mes travaux de recherche.
Mes remerciements vont également au Pr. Olivier Coqueret qui m’a accueilli au sein de son laboratoire. Je le remercie pour la confiance et l’autonomie qu’il m’a accordé durant cette thèse. Merci également pour les conseils et l’aide que tu m’as apportés dans la recherche d’un post-doctorat.
Merci au Dr. Catherine Guette pour sa gentillesse et le temps qu’elle m’a accordé dans la réalisation d’études protéomiques. Je te remercie également pour ta disponibilité et les réponses que tu as apportées à mes nombreuses interrogations.
Merci au Dr. Éric Lelièvre pour ses conseils, sa sympathie, sa bonne humeur et l’aide qu’il a apporté lors de la révision du papier sur CD47.
Merci au Dr. Daniel Pouliquen pour ses encouragements et pour m’avoir donné l’opportunité de participer aux travaux sur S100A4.
Cécile, merci pour ta profonde gentillesse et le temps que tu m’as accordé en cytométrie de flux. Je te remercie également pour tes encouragements et toutes les attentions que tu as eu à mon égard.
Alice, merci pour ta bonne humeur communicative et tes anecdotes hilarantes. Merci également d’avoir pris le temps de me former à la préparation d’échantillons pour une analyse en spectrométrie de masse.
Je tiens à remercier mon professeur de SVT du lycée, Mr. Patrick Ferreira. Tu as été un élément clé dans mon orientation. Tu m’as transmis toute ta passion pour la biologie ainsi que la rigueur et la précision indispensables dans ce domaine.
Raneem, merci pour ta gentillesse et ta bienveillance. Je te remercie également pour tes encouragements. Coralie, merci pour ta bonne humeur et tes encouragements. Je te remercie pour les longues heures passées ensemble devant le cytomètre de flux à analyser l’expression des marqueurs sénescents dans les cellules CD47Low et CD47High, désormais nous savons reconnaitre au premier coup d’œil un problème de fuite ! J’ai
apprécié travailler à tes côtés lors de la révision du papier sur CD47.
Amine, merci pour ta gentillesse et ta volonté d’aider au laboratoire. Je me souviendrai longtemps des fous rires que tu nous as procurés en salle de pause.
Amélie, merci pour ta gentillesse et pour les bons moments passés après le laboratoire. Notre road trip en Irlande sera l’un de mes meilleurs souvenirs, une véritable parenthèse enchantée entre deux années de thèse
Amandine, un merci n’est pas suffisant tant tu m’as apporté durant cette thèse. Tu as été une confidente privilégiée dans les moments difficiles. J’ai apprécié nos nombreuses discussions sur les perspectives de nos travaux. J’ai bien sûr adoré également nos périples sur les routes du GR20 ou bien en Irlande. Ta perpétuelle bonne humeur et ton écoute m’ont vraiment aidé durant cette thèse.
Bertrand, merci pour toute l’aide que tu m’as apportée. Merci pour tout le travail que tu as accompli, tu es le pilier de ce laboratoire ! J’ai adoré travailler à tes côtés durant cette dernière année de thèse. Je te remercie d’avoir pris le temps de me former à la culture d’organoïdes tumoraux. J’ai énormément appris à tes côtés. Nos petits bilans d’organoïdes du matin me manqueront.
Je remercie mes amis de longue date : Julien, Charline, Cloé, Manon, Benoit, Maël et Elodie, les moments passés avec vous m’ont permis de décrocher de mon sujet de thèse et ont été de véritables bulles d’air salvatrices. Je suis heureux que les années passées n’aient pas altérées notre amitié.
Merci Guy, quand tu nous as quitté je ne savais pas encore dans quelle direction je m’orienterai mis à part la science. Avec du recul aujourd’hui, je crois que tu as été pour moi un modèle qui a inconsciemment gouverné mes choix. Quand j’étais plus jeune, tes innombrables connaissances dans le domaine scientifique me fascinaient. Je te revois encore m’expliquer le fonctionnement d’une locomotive à vapeur. Merci pour tous ces moments.
Je remercie ma tante Nelly pour son comportement exemplaire durant cette année difficile. Je te remercie également pour ta disponibilité et ton écoute. Je savais que je pouvais compter sur toi si j’avais besoin d’aide ou de me confier.
Je remercie mes grands-parents (papi Daniel et mamie Jacqueline) pour les coupures du week-end autour d’un café. Ces moments me permettaient d’évacuer la pression et de me changer les idées surtout dans les moments difficiles. Je vous remercie pour votre écoute et vos encouragements.
Je remercie ma sœur Léana pour sa gentillesse et tout le temps qu’elle m’a accordé depuis le début de mes études. Je te sollicitais souvent pour te demander de me faire réciter certaines leçons et tu as toujours répondu présente. J’ai adoré nos petites escapades frère-sœur. Je te remercie également pour ta capacité d’écoute et pour les attentions que tu as eu mon égard. Même si j’étais moins disponible ces derniers temps, je suis fier et heureux de voir que tu as brillamment terminé tes études et que tu t’épanouies pleinement dans ton travail où tu excelles.
Je remercie mes parents pour l’immense soutien qu’ils m’ont porté depuis le début. Je vous remercie de m’avoir donné l’opportunité de suivre la voie qui me plaisait. Vous avez été de véritables moteurs dans les moments de doute. Votre soutien et vos innombrables attentions m’ont permis de trouver l’énergie pour avancer dans mes travaux. Si j’ai eu le bonheur de faire une thèse c’est indéniablement grâce à vous, un simple merci est alors bien modeste tant je vous en suis reconnaissant.
Mamie, je te remercie pour toute l’attention que tu m’as portée. J’ai toujours été fasciné par ton caractère ta volonté et ton courage. J’adorais les innombrables astuces que tu partageais avec nous ainsi que tes anecdotes. Tu étais une véritable Wonder Woman. Tu as été admirable dans la maladie, tu t’es battu tout le temps que cela a été possible. Tu as été un véritable modèle, tu m’as donné l’envie et l’énergie de progresser. Pour toutes ces raisons je te dédie cette thèse.
Table des matières
TABLE DES MATIERES ... 5
LISTE DES ABREVIATIONS : ... 1
INTRODUCTION ... 3
I. La sénescence : un mécanisme pléiotropique ... 5
A. La sénescence réplicative ... 5
1) L’érosion télomérique est à l’origine de la limite réplicative ... 5
2) La sénescence réplicative est associée au vieillissement ... 7
B. La sénescence est induite de manière programmée lors du développement embryonnaire ... 7
1) Les cellules sénescentes sont présentes durant le développement embryonnaire ... 7
2) Régulation de la sénescence développementale ... 8
C. Les cellules sénescentes favorisent le développement de pathologies ... 10
1) La sénescence est impliquée dans le développement de pathologies liées à l’âge ... 10
2) La sénescence favorise le développement de pathologies indépendantes de l’âge ... 12
D. La sénescence se met en place suivant la surexpression d’oncogènes ... 13
1) La surexpression d’oncogènes induit une entrée en sénescence ... 13
2) Les dommages de l’ADN sont responsables de l’entrée en sénescence oncogénique ... 13
3) La sénescence oncogénique limite la formation de tumeurs malignes in vivo ... 14
E. La sénescence chimiothérapeutique bloque la prolifération des cellules tumorales en réponse aux traitements génotoxiques. ... 16
1) Les traitements génotoxiques induisent une entrée en sénescence ... 16
2) Régulation du choix apoptose-sénescence suivant un traitement génotoxique ... 17
2a Le niveau de dommages de l’ADN module le choix apoptose-sénescence : ... 17
2b L’activation d’oncogènes comme AKT et la perte de suppresseurs de tumeur tels que PTEN modulent le choix apoptose-sénescence : ... 19
3) La dernière génération de thérapies anti-cancéreuses induit une entrée en sénescence ... 19
II. Caractéristiques cellulaires et moléculaires des cellules sénescentes ... 22
A. Arrêt du cycle cellulaire : ... 22
1) Régulation de l’expression des inhibiteurs du cycle cellulaire p21 et p16. ... 22
2) Inhibition de la prolifération par le blocage de l’activité des complexes Cycline/Cdk : Rôle de p21 et p16 ... 24
3) Maintien de l’arrêt prolifératif : Activation de pRb ... 25
B. Remodelage chromatinien : épigénétique et irréversibilité de l’arrêt prolifératif. ... 26
1) Répression épigénétique des gènes prolifératifs par Rb et formation de SAHFs ... 26
2) Régulation de la formation des SAHFs ... 27
3) Les cellules sénescentes sont caractérisées par la persistance de dommages de l’ADN : DNA-SCARS. ... 28
4) Réorganisation générale de la chromatine au sein des cellules sénescentes ... 28
C. L’entrée en sénescence s’accompagne d’importantes modifications morphologiques ... 30
1) Hypertrophie des cellules sénescentes ... 30
2) Dérégulation de l’activité autophagique : dégradation de l’enveloppe nucléaire et activité SA- β-galactosidase ... 31
3) Modifications morphologiques des mitochondries ... 32
D. Reprogrammation métabolique ... 33
1) Modification de l’activité glycolytique des cellules sénescentes... 33
2) Les oncogènes tels que Ras augmentent l’activité glycolytique des cellules sénescentes... 34
3) Augmentation de la concentration calcique mitochondriale au sein des cellules sénescentes ... 34
E. Résistance à l’apoptose et dépendance aux signaux pro-survie : Intérêt des sénolytiques. ... 35
1) Les cellules sénescentes sont intrinsèquement résistantes aux stimuli apoptotiques : p21 inhibiteur du processus apoptotique ... 35
2) La viabilité des cellules sénescentes dépend de signaux pro-survie : intérêt des sénolytiques ... 36
F. Les cellules sénescentes sont caractérisées par un déséquilibre protéique ... 38
1) Les cellules sénescentes arborent un stress protéique : rôle des acteurs de l’UPR ... 38
1) Les cellules sénescentes développent un phénotype sécrétoire : Rôle de NF-κB et C/EBP-β ... 40
2) Les dommages de l’ADN sont nécessaires à l’établissement du SASP : ... 42
3) Régulateurs additionnels du SASP : ... 44
4) La composition du SASP est modulée par le métabolisme, l’expression de suppresseurs de tumeur comme p53, d’oncogènes tels que Ras et l’activité de récepteurs comme NOTCH1. ... 45
III. La sénescence est un acteur de la transformation et de la progression tumorale : un mécanisme d’adaptation responsable de l’échec thérapeutique ? ... 47
A. La sénescence régule l’immunité anti-tumorale. ... 47
1) La fonction suppressive des cellules sénescentes requiert leur élimination par le système immunitaire ... 47
2) L’élimination des cellules sénescentes est régulée par l’expression de facteurs spécifiques ... 48
3) Le recrutement de cellules immunitaires suppressives par les cellules sénescentes abolit la surveillance immunitaire 50 B. Les cellules sénescentes augmentent l’agressivité des cellules tumorales ... 52
1) Les cellules sénescentes favorisent la croissance tumorale : Augmentation du processus angiogénique et de la prolifération des cellules cancéreuses. ... 52
2) La dualité de fonction du SASP est contrôlée par l’activité de kinases telles que mTOR, de protéines suppressives comme p53 et par l’oncogène Ras. ... 53
3) Les cellules sénescentes favorisent la formation de métastases : ... 53
4) Les cellules sénescentes favorisent la reprogrammation et la sélection de cellules souches. ... 54
C. L’échappement à la sénescence réplicative favorise la transformation ... 56
1) Les mécanismes de stress assurent la pérennité de l’état sénescent ... 56
2) Les cellules tumorales surexpriment les acteurs permettant l’échappement à la sénescence réplicative ... 57
D. L’échappement à la sénescence oncogénique favorise la transformation cellulaire ... 58
1) L’expression d’oncogènes secondaires comme Myc, la coopération avec des protéines intracellulaires telles que Rac1b et les facteurs sécrétés favorisent l’échappement à la sénescence oncogénique ... 58
2) La perte d’expression de protéines suppressives de tumeur telles que p16 ou PTEN favorise l’échappement à l’OIS 60 3) L’échappement à la sénescence oncogénique est favorisé par l’expression de facteurs de l’EMT et est associé à l’expression de facteurs souches. ... 60
E. L’échappement à la sénescence chimiothérapeutique est responsable de l’échec thérapeutique ... 62
1) Des cellules échappent à la sénescence induite par les thérapies anti-cancéreuses ... 62
2) L’échappement à la sénescence chimiothérapeutique est favorisé par l’expression d’oncogènes tels que Src et de kinases comme AKT ... 64
3) La surexpression de p21 génère une instabilité génétique favorisant l’échappement à la chimiothérapie ... 65
4) L’échappement à la sénescence chimiothérapeutique favorise la reprogrammation des cellules tumorales et l’émergence de cellules au phénotype souche... 66
F. Conclusion générale ... 68
RESULTATS ... 73
DISCUSSION ET PERSPECTIVES ... 173
IV. L’hétérogénéité de la sénescence est définie au niveau membranaire et dépend de l’activité d’endocytose ? . 175 V. L’hétérogénéité de la sénescence est générée au niveau transcriptionnel : Rôle de Myc et de ses partenaires transcriptionnels ? ... 177
VI. L’hétérogénéité de la sénescence est définie au niveau traductionnel : la biosynthèse d’ARN de transfert au cœur de la suppression tumorale ? ... 179
A. L’activité de l’ARN Polymérase III module la stabilité de l’état sénescent et contribue à la progression tumorale. . 179
B. La surexpression d’ARN de transfert spécifiques favorise la progression tumorale ? ... 181
C. Les activités détournées de la traduction des ARNt-ligases perturbent l’activation des voies oncogéniques et suppressives. ... 184
D. La dérégulation de la synthèse d’ARN de transfert génère une diversité protéique au cœur de la progression tumorale ? 186 BIBLIOGRAPHIE ... 190
TABLE DES FIGURES ... 203
Liste des abréviations :
ADN : Acide désoxyribonucléique ARN : Acide ribonucléique
ATF6 : Activating transcription factor 6 ATM : Ataxia telangiectasia mutated
ATR : Ataxia telangiectasia and Rad3-related protein BRDU : Bromodésoxyuridine
CCF : Cytoplasmic chromatin fragments Cdk : Cyclin-dependent kinase
Cdki : Cyclin-dependent kinase inhibitor DDR : DNA damage response
DFO : Déféroxamine
DNA-SCARS : DNA segments with chromatin alterations reinforcing senescence EGFR : Epithelial growth factor receptor
EZH2 : Enhancer of zeste homolog 2
H3K27me3 : Tri-méthylation de la lysine 27 de l’histone H3 H3K9me3 : Tri-méthylation de la lysine 9 de l’Histone H3 HGF : Hepatocyte growth factor
HMGA : High mobility group AT-hook protein ITPR2 : Inositol 1,4,5-trisphosphate receptor type 2 JAK2 : Janus kinase 2
MCM : Minichromosome maintenance protein MDSC : Myeloid-derived suppressor cells mTOR : Mammalian target of rapamycin NAD : Nicotinamide adenine denucleotide NK : Natural Killer
OIS : Oncogene-induced Senescence PAI-1 : Plasminogen activator inhibitor 1 PCNA : Proliferating cell nuclear antigen PDGF : Platelet-derived growth factor
PERK : PRKR-like endoplasmic reticulum kinase PI3K : Phosphatidylinositol-3 kinase
PIN : Néoplasie intraépithéliale prostatique PKC : Protein kinase C
PRC1 : Polycomb Repressive Complexe 1 PRC2 : Polycomb Repressive Complexe 2 ROS : Reactive oxygen species
SAHF : Senescence-associated heterochromatin foci SASP : Senescence-associated secretory phenotype
SA-β galactosidase : Senescence-associated β-galactosidase SMS : Senescence messaging secretome
STAT3 : Signal transducer and activator of transcription 3 TGFβ : Transforming growth factor-beta
TSP1 : Thrombospondin-1 UPR : Unfolded protein response
VEGF : Vascular endothelial growth factor ZFP36L1 : Zinc finger protein 36, C3H1-like 1
I. La sénescence : un mécanisme pléiotropique
A. La sénescence réplicative
1) L’érosion télomérique est à l’origine de la limite réplicative
Le concept de sénescence fut abordé pour la première fois par Hayflick and Moorhead en 1961. Ils observèrent que les cellules primaires humaines adultes stoppaient définitivement leur prolifération après environ 50 passages en culture 1,2. Plus tardivement, l’analyse des extrémités chromosomiques, les télomères,
a permis de comprendre l’origine de la limite réplicative dénommée « limite de Hayflick ». L’étude de Harley et al a mis en évidence que la taille des télomères était progressivement réduite au cours des divisions successives. A l’inverse, la quantité d’autres séquences répétées non télomériques n’est pas modifiée. Ceci indique que la réduction de la taille des télomères n’est pas la conséquence d’une dégradation générale de l’ADN mais qu’elle est restreinte à cette région du génome 3. Le raccourcissement télomérique est dû au
processus réplicatif semi-conservatif. En effet, l’ADN polymérase fonctionne uniquement dans le sens 5’ à 3’. Elle nécessite ainsi d’une amorce d’ARN pour répliquer l’extrémité 3’ libre. Ceci conduit alors à une perte progressive de l’extrémité 3’ qui ne peut être totalement répliquée 4. Les télomères se composent d’une
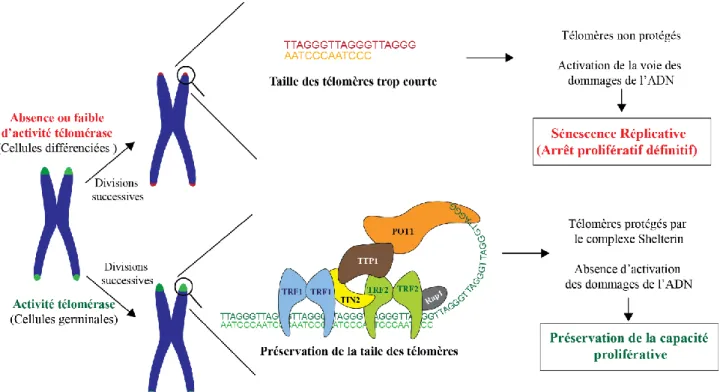
répétition en tandem de la séquence TTAGGG chez l’Homme. Elle est directement reconnue par les protéines TRF1, TRF2 et POT1 du complexe Shelterin. Ces protéines associées aux trois autres membres de ce complexe, TIN2, TTP1 et Rap1, régulent la taille des télomères. Elles empêchent la fusion des extrémités chromosomiques libres et ainsi l’activation de la voie des dommages de l’ADN 5. Dès lors, la surexpression
d’un dominant négatif de TRF2 cause la fusion des extrémités télomériques et induit l’entrée en sénescence des cellules 6.
Ainsi, l’intégrité télomérique doit être conservée afin de maintenir la capacité proliférative des cellules. Celle-ci est régulée par une ribonucléoprotéine, la télomérase. Cette enzyme est constituée de deux sous-unités, une sous-unité ARN nommée hTERC et une sous-unité catalytique hTERT. Elle fonctionne comme une réverse transcriptase et produit les répétitions télomériques en tandem à partir de la séquence d’ARN donneur fournie par hTERC 7. Cependant, l’expression de la télomérase est quasiment indétectable
dans la plupart des cellules somatiques chez l’adulte 8. Ceci est la cause de la capacité réplicative limitée des
cellules différenciées adultes. En l’absence de cette enzyme, la taille des télomères est alors progressivement réduite au cours des divisions jusqu’à une longueur critique favorisant la fusion des chromosomes et une entrée en sénescence 9. L’étude de Fabrizio d’Adda di Fagagna et al met en évidence par la culture de fibroblastes
tels que γH2AX et 53BP1 au niveau des télomères. A l’inverse, les cellules en phase proliférative ou en quiescence n’expriment que très faiblement ces marqueurs. Les cellules en sénescence réplicative activent donc la voie des dommages de l’ADN comme illustré par l’activation des kinases ATM, Chk1 et Chk2. L’érosion des télomères favorise donc l’accumulation de dommages de l’ADN. Ainsi, la surexpression du dominant négatif de la protéine TRF2 induit l’activation de cette voie en plus de promouvoir l’entrée en sénescence. La déplétion des kinases ATM, Chk1 ou Chk2 dans les fibroblastes sénescents augmente l’expression du marqueur prolifératif Mcm7 et l’incorporation de BRDU 10. A l’inverse, la surexpression de
la télomérase dans des fibroblastes humains diploïdes réduit les cassures ce qui empêche leur entrée en sénescence et induit ainsi leur immortalisation 11.
Ainsi, l’érosion télomérique est directement associée à la voie des dommages de l’ADN dont l’activation est indispensable à l’entrée en sénescence réplicative. Les premières études étant effectuées in
vitro, la validation de l’existence de ces cellules in vivo a été effectuée dans un second temps (Figure 1).
Figure 1 : Mécanismes responsables de la maintenance de la capacité proliférative ou de l’entrée en sénescence réplicative.
Dans les cellules différenciées adultes, en l’absence d’activité télomérase, la taille des télomères est progressive réduite aboutissant à l’activation de la voie des dommages de l’ADN et leur entrée en sénescence réplicative. La télomérase en association avec les membres du complexe Shelterin maintient l’intégrité des extrémités télomériques préservant ainsi la capacité proliférative des cellules.
2) La sénescence réplicative est associée au vieillissement
La sénescence réplicative a ainsi été retrouvée in vivo. En effet, les babouins âgés ont une augmentation du pourcentage de cellules de la peau présentant une dysfonction de leurs télomères en comparaison aux membres plus jeunes. Ces cellules expriment les marqueurs témoignant de l’activation des dommages de l’ADN tels que 53BP1. Le pourcentage de cellules présentant un dysfonctionnement des télomères est alors associé à l’expression de p16 qui est nécessaire au maintien de la sénescence 12. La
sénescence réplicative a également été retrouvée chez l’Homme suite à l’étude de l’activité β-galactosidase associée à la sénescence (SA-β galactosidase) dans des cellules du derme et de l’épiderme de patients jeunes et âgés. Les jeunes patients ont une activité SA-β galactosidase très faible voire absente alors que cette activité est importante dans le derme et l’épiderme de patients âgés. Ceci indique que les cellules sénescentes s’accumulent in vivo chez l’Homme au cours du vieillissement 13.
Ainsi, la sénescence réplicative constitue une limite proliférative des cellules adultes. Elle est induite par l’érosion progressive des extrémités chromosomiques, les télomères, ce qui entraine l’activation de la voie des dommages de l’ADN. Ce type de sénescence n’est pas un artéfact de culture in vitro puisqu’il est retrouvé
in vivo chez l’Homme.
B. La sénescence est induite de manière programmée lors du développement embryonnaire
1) Les cellules sénescentes sont présentes durant le développement embryonnaire
La sénescence n’est pas uniquement associée au vieillissement cellulaire. En effet, la présence de cellules sénescentes a pu être observée au niveau d’embryons de poulet et de souris. Ces cellules ont été identifiées après marquage des embryons à l’activité SA-β galactosidase. Elles sont localisées sur des zones précises des embryons, au niveau de la crête apicale ectodermale et du sommet de la plaque neurale. De plus, ces cellules apparaissent et disparaissent à des stades précis lors du développement embryonnaire. Ces résultats indiquent que la sénescence se met en place de manière programmée durant le développement embryonnaire
14. Leur présence a également été retrouvée dans l’étude de Muñoz-Espín et al y compris chez l’homme au
niveau du sac endolymphatique et du mésonéphros. Les cellules sénescentes observées expriment principalement l’inhibiteur du cycle cellulaire p21 mais aussi p15 et p27. A l’inverse, l’expression du suppresseur de tumeur p53 est très faible voire indétectable 15. Plus récemment, la sénescence a également été
observée durant le développement embryonnaire chez l’amphibien. Les cellules sénescentes participent à la dégénérescence physiologique du pronéphros et au développement des cavités orales 16.
2) Régulation de la sénescence développementale
Contrairement à la sénescence réplicative, ce type de sénescence est indépendante des dommages de l’ADN. Les embryons déficients en ATM ou ATR ne présentent pas d’altérations du nombre de cellules exprimant l’activité SA-β galactosidase au niveau du mésonephros et du sac endolymphatique. La sénescence développementale est à l’inverse dépendante de p21 mais pas des suppresseurs de tumeur p53 et p16. Par comparaison des tubules mésonéphrétiques d’embryons normaux ou déficients pour l’expression de p21, il a été observé que les ARN liés aux voies développementales TGFβ, Hedgehog et WNT sont plus fortement exprimés dans les embryons sauvages. Plus particulièrement, le rôle de la voie TGFβ/SMAD a été étudié puisqu’elle est capable d’activer la transcription de p21. Au niveau des cellules épithéliales des tubules mésonéphrétiques et du sac endolymphatique l’expression de SMAD2 phosphorylée a pu être détectée. Afin d’identifier la fonction de la voie TGFβ, l’utilisation de l’inhibiteur (LY2152799) sélectif pour le principal récepteur du TGFβ, TGFBR1, a été utilisé par gavage oral quotidiennement sur des souris gestantes entre le stade embryonnaire E10.5 et E14.5. En plus, de la réduction de la phosphorylation activatrice de SMAD2 le nombre de cellules SA-β galactosidase positives et l’expression de p21 est significativement réduite. Ainsi, le TGFβ est indispensable à l’entrée en sénescence développementale. L’expression de p21 pouvant également être régulée par les protéines FOXO, les auteurs ont étudié l’expression de la forme phosphorylée inactive de FOXO et celle de cet inhibiteur du cycle cellulaire par immunofluorescence. De manière intéressante, les cellules exprimant fortement la forme inactive de FOXO1 ou 3 expriment faiblement p21. L’utilisation d’inhibiteurs de la kinase PI3K permet d’accroître l’activité de FOXO. En conséquence, l’activité SA-β galactosidase et l’expression de p21 est augmentée sans modifications du nombre de cellules exprimant cet inhibiteur du cycle cellulaire. Ainsi, en plus de la voie du TGFβ, FOXO serait nécessaire au renforcement de l’état sénescent par l’augmentation de l’expression de p2115.
Les auteurs ont ensuite étudié le développement d’embryons déficients pour l’expression de p21 ne pouvant accumuler des cellules sénescentes. Au niveau de la crête ectodermale d’embryons, l’absence de p21 est associée à l’augmentation du nombre de cellules mortes. De plus, ces embryons ont une réduction de la prolifération du mésenchyme au-dessous de la crête ectodermale. Ceci est expliqué par un défaut d’expression des inducteurs de la prolifération issue de la crête ectodermale apicale, FGF4 et FGF8 14. Par ailleurs, lors du développement embryonnaire, les cellules sénescentes pourraient participer à l’élimination de certaines
présentes au stade embryonnaire E14.5 d’embryons murins et sont entourées de macrophages. Plus tardivement, entre le stade embryonnaire E15.5 et 16.5, les tubules caudaux sont éliminés alors que les macrophages sont toujours présents. D’une manière générale, un défaut d’entrée en sénescence durant le développement embryonnaire semble compensé par l’induction de l’apoptose qui permet la régression du tissu. Cependant, au niveau de certaines zones anatomiques, ce défaut est associé à des malformations lors du développement ayant des conséquences à l’âge adulte. Ceci, est illustré par la comparaison des canaux de Wolf entre des souris mâles et femelles. En effet, dans le cas des femelles et contrairement aux mâles, les canaux de Wolf régressent. Ceci est corrélé à une différence d’expression de l’activité SA-β galactosidase. Cette activité est forte pour les femelles alors qu’elle est quasiment indétectable chez les souris mâles. La perte de p21 est alors associée à une augmentation de la fréquence d’apparition de souris possédant de manière anormale un septum dorsoventral au niveau du vagin. Cette anomalie est observée dans plus de 15% des souries déficientes pour l’expression de p21. Naturellement, elle est retrouvée dans 3.75% des souris sauvages et engendre une réduction de la fertilité 15.
La présence de cellules sénescentes chez l’Homme a également été retrouvée durant l’involution thymique se produisant peu de temps avant la puberté. Les cellules sénescentes se situent à proximité des corpuscules de Hassal uniquement chez les sujets adolescents (11-14 ans) et chez les sujets plus âgés (34-73 ans). Chez les sujets adolescents, les cellules sénescentes n’expriment pas les marqueurs d’activation de dommages de l’ADN contrairement aux sujets plus âgés. Ainsi, une sénescence indépendante des dommages de l’ADN se met également en place chez l’Homme lors de l’involution thymique 17.
La sénescence développementale participe donc au remodelage tissulaire durant le développement embryonnaire. Elle dépend de l’activité du TGFβ et de FOXO nécessaires à l’expression de p21. La sénescence favorise la croissance ou bien l’élimination de certaines structures durant le développement embryonnaire (Figure 2).
Figure 2 : Voies impliquées dans la régulation de la sénescence développementale.
L’expression de p21 est régulée positivement par la voie TGFβ/SMAD qui favorise son expression et l’entrée en sénescence. L’activation de FOXO favorise le renforcement de l’état sénescent en augmentant l’expression de p21. La sénescence induite de manière programmée favorise le remodelage tissulaire durant le développement embryonnaire. Adapté de Daniel Muñoz-Espín et al, Cell, 2013 15.
C. Les cellules sénescentes favorisent le développement de pathologies
1) La sénescence est impliquée dans le développement de pathologies liées à l’âge
Malgré que la sénescence participe au remodelage tissulaire lors du développement embryonnaire. Elle peut aussi favoriser la survenue de certaines pathologies lors du vieillissement. Cette fonction n’a été appréhendée que très récemment par les travaux de Baker et al. Dans cette étude, les auteurs ont utilisé le modèle murin transgénique INK-ATTAC exprimant la protéine de fusion FK506-binding-protein-caspase 8 et la protéine fluorescente GFP sous le contrôle du promoteur de p16. Ce système permet la visualisation des cellules sénescentes et leur élimination spécifique par l’administration de l’AP20187 permettant l’activation de la protéine de fusion FKBP-Caspase 8. Par l’utilisation de ce système, les auteurs ont pu confirmer l’accumulation de cellules sénescentes dans les différents tissus au cours du vieillissement. Leur élimination chez les souris âgées de 12 mois accroit leur survie. Elle augmente également celle de souris développant des tumeurs, cependant, l’élimination des cellules sénescentes ne réduit pas l’occurrence de cancers mais limite leur développement. Ces cellules détectées au niveau des différents tissus sont impliquées dans le développement de pathologies liées à l’âge. En effet, leur élimination retarde l’apparition de la cataracte, de la lipodystrophie, de l’hypertrophie des cardiomyocytes et de la glomérulosclérose. L’implication de ces cellules dans la survenue de diverses pathologies liées au vieillissement explique pourquoi leur élimination augmente l’espérance de vie en bonne santé des modèles murins étudiés 18.
Afin de valider le rôle effectif des cellules sénescentes dans le vieillissement, dans une autre étude, les auteurs utilisent un modèle murin chez lequel ils récupèrent des cellules saines qu’ils rendent ou non sénescentes par irradiation ou bien par traitement à la chimiothérapie. Ils les injectent ensuite dans des souris jeunes âgées de 6 mois ou bien vieilles âgées de 17 mois. L’injection de cellules sénescentes diminue leurs capacités physiques après seulement 1 mois et réduit également leur espérance de vie. Cependant, de manière cohérente avec l’étude précédente, elle n’augmente pas le risque de formation de tumeurs. Le lien entre l’injection de cellules sénescentes et la réduction des capacités physiques a été vérifié par l’utilisation de drogues sénolytiques. Leur utilisation limite la perte d’activité physique causée par les cellules sénescentes. De plus, l’administration de ces agents entre le 20ième et 24ième mois de vie des souris augmente
significativement leurs capacités physiques et leur espérance de vie 19.
Les cellules sénescentes sont également impliquées dans la dégradation du tissu osseux. En effet, dans le modèle transgénique INK-ATTAC ou par l’utilisation de drogues sénolytiques, il a été observé que l’élimination des cellules sénescentes dans des souris âgées présentant une perte osseuse avérée augmente la masse, la force ainsique la microarchitecture osseuse. En sécrétant des facteurs tels que PAI-1, 6 et l’IL-8, les cellules sénescentes favorisent la survie des progéniteurs ostéoclastogéniques et ainsi l’ostéoclastéogénèse, c’est-à-dire la dégradation osseuse. A l’inverse, elles limitent la minéralisation osseuse par les ostéoblastes 20. Leur présence a été détectée au niveau du cartilage articulaire et du synovium de souris
âgées ayant subi une opération suite à une déchirure des ligaments croisés. Leur élimination par l’utilisation des modèles murins transgéniques INK-ATTAC ou p16-3MR limite le développement d’arthtose post-traumatique ainsi que la douleur associée et augmente la formation de cartilage. La présence de chondrocytes sénescents a également été identifiée sur des d’échantillons de patients souffrants d’arthrose et ayant subi une opération du genou. La culture in vitro des chrondocytesissus de patients a montré que ces cellules sécrètent des facteurs pro inflammatoires tels que l’IL-1β et l’IL-6 et à l’inverse limitent l’expression de protéines de la matrice extracellulaire du tissu cartilagineux comme COL2A1 et ACAN 21.
Enfin, les cellules sénescentes sont également associées à certaines maladies neurodégénératives. Effectivement, dans le modèle murin MAPTP301SPS19 de maladie neurodégénérative, l’accumulation
d’astrocytes et de microglie sénescente a été identifiée. Leur élimination par l’utilisation du modèle murin INK-ATTAC réduit la gliose et l’hyperphosphorylation des protéines tau insolubles, limitant ainsi la dégénération des neurones corticaux et hippocampaux. L’élimination des cellules sénescentes permet dès lors le maintien des capacités cognitives telles que la discrimination d’objets 22. De même, l’étude de tissus du
cortex frontal de patients âgés a mis en évidence que le nombre d’astrocytes exprimant p16 est significativement plus important chez les patients souffrants d’Alzheimer que ceux d’âges similaires non
atteints par cette pathologie 23. L’accumulation de cellules sénescentes au niveau du cerveau pourrait donc
favoriser le développement de pathologies cognitives.
Ainsi, l’ensemble des études précédentes démontrent que les cellules sénescentes participent à l’établissement de nombreuses pathologies liées au vieillissement. Ceci réduit l’espérance de vie dans les modèles murins étudiés.
2) La sénescence favorise le développement de pathologies indépendantes de l’âge
Les cellules sénescentes ne sont pas seulement impliquées dans la promotion de pathologies liées à l’âge, elles participent également à la survenue de pathologies indépendantes de celui-ci. Effectivement, dans un modèle murin d’obésité induit soit par un régime riche en graisse soit par l’inactivation du récepteur à la leptine, l’accumulation de cellules sénescentes gliales est détectée au niveau du ventricule latéral. A l’inverse, les souris non obèses ne présentent pas d’accumulation de cellules sénescentes au niveau de cette région anatomique. L’obésité est souvent associée à des troubles comportementaux comme l’anxiété et la dépression. L’élimination des cellules sénescentes diminue significativement l’anxiété associée à cette pathologie. De plus, elle restaure la neurogénèse habituellement réduite dans les souris obèses 24. Par ailleurs, la présence de
cellules sénescentes a aussi été identifiée au niveau de plaques d’athérome dans le modèle murin transgénique p16-3MR. Dans ce modèle, l’utilisation du Ganciclovir permet l’élimination des cellules sénescentes exprimant p16. Par l’utilisation de ce système, il a été démontré que les cellules sénescentes participent à la formation et l’augmentation de la taille des plaques d’athérome 25.
Les cellules sénescentes favorisent donc le développement de pathologies indépendantes de l’âge. Cependant, dans certains cas, les cellules sénescentes limitent le développement de pathologies ou bien leur ampleur. Dans le cas de la fibrose hépatique, l’accumulation de cellules sénescentes diminue la propagation de cellules de Kupffer et la formation d’une fibrose excessive. Elles réduisent la sécrétion de composants de la matrice extracellulaire et à l’inverse augmentent celle d’enzymes de dégradation de la matrice. De plus, elles favorisent la surveillance immunitaire par les cellules Natural Killer (NK) facilitant ainsi la résolution de la fibrose 26.
Ainsi, les cellules sénescentes s’accumulent dans de nombreux tissus au cours du vieillissement. Elles favorisent alors le développement de pathologies liées à l’âge mais également non associées à celui-ci. Dans de plus rares cas, comme la fibrose hépatique, elles limitent l’ampleur de la maladie. Cependant, d’une manière générale, leur élimination augmente la viabilité dans les modèles murins étudiés.
D. La sénescence se met en place suivant la surexpression d’oncogènes
1) La surexpression d’oncogènes induit une entrée en sénescence
Un nouveau type de sénescence fut découvert par Serrano en 1997. En effet, il observa que la surexpression de la forme oncogénique de H-Ras (H-RasV12) dans des fibroblastes primaires murins et humains
n’induisait pas leur transformation mais bloquait définitivement leur prolifération de manière dépendante de l’expression de p53 et p16. Cet arrêt prolifératif et les changements morphologiques furent associés à l’entrée en sénescence du fait que les fibroblastes avaient une activité SA-β galactosidase 27. Cette sénescence induite par les oncogènes est maintenant appelée sénescence oncogénique (Oncogene-Induced Senescence : OIS). Elle est retrouvée dans de multiples études et a pu être généralisée avec d’autres oncogènes tels que STAT5A ou bien le proto-oncogène Mos. Effectivement, la surexpression de ces oncogènes bloque la prolifération et promeut le développement d’une activité SA-β galactosidase de fibroblastes humains 28,29. Cependant, tous les
oncogènes n’induisent pas une entrée en sénescence. Par exemple, la surexpression de l’oncogène Myc induit une entrée en apoptose de fibroblastes primaires 30. Parfois, c’est la perte d’expression d’une protéine suppressive de tumeur qui est responsable de l’activation de ce mécanisme. En effet, l’inactivation de PTEN induit l’entrée en sénescence de fibroblastes primaires 31. Ce type de sénescence n’est pas la conséquence de
conditions particulières de culture in vitro ne reflétant pas la réalité in vivo. Effectivement, la présence de cellules sénescentes sur des échantillons de nævi humains exprimant la forme oncogénique de B-Raf (B-RafV600E) a été observée. Les marquages immunohistochimiques révèlent que les nævi expriment les
marqueurs sénescents tels que l’inhibiteur du cycle cellulaire p16, l’activité SA-β galactosidase et qu’ils perdent l’expression de l’antigène prolifératif Ki67 32.
Ainsi, la surexpression d’oncogènes ou bien la perte de protéines suppressives de tumeur induisent in
vitro et in vivo l’entrée en sénescence de cellules primaires.
2) Les dommages de l’ADN sont responsables de l’entrée en sénescence oncogénique
L’analyse de ce type de sénescence a permis d’identifier la nécessité de l’activation de la voie des dommages de l’ADN pour son établissement. Dans un premier temps, il a été observé que les cellules en sénescence oncogénique médiée par Ras expriment les formes phosphorylées et actives des acteurs de cette voie tels que Chk2 (T68), de Chk1 (S345), d’ATM (S1981) et p53. Leur activation est la conséquence d’un stress réplicatif consécutif à l’augmentation de la prolifération dans les premiers jours suivant la surexpression de Ras. Durant ce laps de temps, l’expression de la protéine réplicative CDC6 augmente ce qui accroit le nombre d’origines de réplication. L’augmentation de l’activité réplicative est la source des dommages de
l’ADN accumulés. En effet, l’inhibition de l’ADN polymérase par l’Aphidicoline bloque l’accumulation des cassures dans les cellules surexprimant la forme oncogénique de Ras.
L’activation de cette voie est indispensable à l’entrée en sénescence oncogénique. La déplétion de Chk2 empêche en effet l’entrée en sénescence des fibroblastes primaires qui surexpriment Ras. Cependant, ces cellules accumulent toujours des dommages de l’ADN. Par ailleurs, la perte de cette kinase permet la transformation des cellules, celles-ci acquièrent la capacité de croitre en conditions de faible adhérence et de former des tumeurs dans des souris immunodéprimées 33. La déplétion d’autres acteurs de la voie des
dommages de l’ADN tels qu’ATM ou p53 bloque également l’entrée en sénescence suivant la surexpression de RasV12, de STAT5A ou suivant la perte de PTEN 28,31,33. Ainsi, quel que soit l’oncogène activé, l’induction
des dommages de l’ADN est indispensable à l’entrée en sénescence. De plus, il semblerait qu’elle soit également nécessaire à son maintien. Effectivement, après la surexpression de RasV12, la déplétion d’ATM,
ATR ou Chk2 induit une reprise de la prolifération 33. L’importance de cette voie est également retrouvée in
vivo par l’étude de coupes de vessies humaines à différents stades tumoraux. Aux stades les plus précoces,
l’expression de marqueurs d’activation de dommages de l’ADN est observée et leur expression est plus forte qu’au niveau de l’épithélium sain. En effet, sur les lésions pré-cancéreuses, la présence des formes phosphorylées actives de Chk2, ATM et p53 est observée. A l’inverse, aux stades plus avancés leur expression est significativement réduite 34. L’activation de la voie des dommages de l’ADN au niveau de lésions
pré-cancéreuses est également associée à l’expression de marqueurs sénescents sur des coupes d’adénome de côlons humains29.
Ainsi, l’établissement de la sénescence oncogénique est associée in vitro et in vivo à l’accumulation de dommages de l’ADN nécessaires à sa mise en place et son maintien.
3) La sénescence oncogénique limite la formation de tumeurs malignes in vivo
La fonction de ce type de sénescence a été étudiée notamment par l’étude d’un modèle murin surexprimant l’oncogène Ras spécifiquement au niveau de l’intestin. La surexpression de cet oncogène induit le développement de lésions hyperplasiques intestinales exprimant les marqueurs sénescents. Lorsque ces souris sont déficientes pour l’expression de p16INK4A, le développement de tumeurs intestinales surexprimant
Ras et capables de former des métastases est constaté. Chez l’Homme, l’expression de p16 au niveau de tumeurs colorectales invasives est significativement réduite au niveau des fronts infiltrants. Par ailleurs, son expression est inversement corrélée à celle de l’antigène prolifératif Ki67. De plus, l’expression de p16 est totalement perdue au niveau d’une tumeur avancée d’adénocarcinome ayant formée des métastases 35. Ainsi,
tels que Ras. Dans le modèle murin transgénique Eµ-N-Ras, les souris hétérozygotes développent des lymphomes T consécutivement à la perte d’expression de Suv39h1 ou de p53. A l’inverse, les souris homozygotes pour ces gènes développent seulement une néoplasie non lymphoïde plus tardive. Chez ces souris, la prolifération des lymphocytes primaires est bloquée par Suv39h1 et ces cellules expriment les marqueurs sénescents 36. Dès lors, l’expression d’acteurs nécessaires à l’entrée en sénescence limite
directement le développement de tumeurs liquides comme un lymphome T.
Dans un autre modèle murin, la perte d’expression de PTEN au niveau de la prostate conduit à la formation de cancers non létaux après une longue période de latence. Ces lésions expriment des marqueurs sénescents tels que l’activité SA-β galactosidase, p53 et p21. Lorsque l’inactivation de PTEN est associée à la perte de p53, les souris développent des tumeurs prostatiques invasives deux semaines après la puberté et décèdent inévitablement à 7 mois. A ce même temps, toutes les souris seulement déficientes pour l’expression de PTEN sont vivantes. Ceci est associé à une diminution drastique des marqueurs sénescents au niveau des tumeurs arborant la double déplétion de PTEN et de p53. Ce phénomène semble retrouvé in vivo chez l’Homme au niveau de lésions prostatiques. En effet, l’activité SA-β galactosidase est très forte chez un patient présentant une lésion pré-néoplasique alors qu’elle est faible chez un autre patient présentant une lésion ayant progressée 31.
Ainsi, la sénescence oncogénique constitue un réel mécanisme de suppression tumorale in vitro et in
vivo y compris chez l’Homme. Elle se met place en suivant l’activation de certains oncogènes ou bien la perte
de suppresseurs de tumeur. Comme la sénescence réplicative, elle est induite par l’activation de la voie des dommages de l’ADN et dépend de l’expression de p53 et p16 (Figure 3).
Figure 3 : Mécanismes responsables de la mise en place de la sénescence oncogénique.
Suivant la surexpression d’oncogènes ou bien la perte de protéines suppressives de tumeur, les cellules ont une activité proliférative accrue qui génère un stress réplicatif. En conséquence, les cellules entrent en sénescence oncogénique (OIS) suivant l’activation de la voie des dommages de l’ADN. Un défaut d’activation de cette voie empêche l’établissement de ce mécanisme suppressif et conduit à la formation de tumeurs malignes.
E. La sénescence chimiothérapeutique bloque la prolifération des cellules tumorales en réponse aux traitements génotoxiques.
1) Les traitements génotoxiques induisent une entrée en sénescence
La sénescence est également impliquée dans la réponse aux agents anti-cancéreux génotoxiques. En effet, de manière similaire à la limite réplicative ou à la surexpression d’oncogènes, les cassures de l’ADN générées par ces agents induisent une entrée en sénescence. Par exemple, le traitement de lignées tumorales d’adénocarcinome colorectal avec le métabolite actif de l’Irinotécan, le Sn38, induit l’établissement de ce mécanisme de suppression tumorale. Les cellules traitées stoppent leur prolifération et expriment des marqueurs sénescents comme l’activité SA-β galactosidase, p53 et p21 37. Ce phénomène est aussi observé
dans les cellules de la lignée de carcinome nasopharyngé, CNE1 traitées au Cisplatine 38. Ce type de suppression tumorale est appelé Therapy-induced Senescence (TIS) ou bien Chemotherapy-induced Senescence (CIS). Cet aspect est également retrouvé in vivo. Effectivement, une tumeur pulmonaire d’un patient ayant subi 3 cycles de chimiothérapie au Carboplatine et Taxol avant l’exérèse de la tumeur résiduelle exprime des marqueurs sénescents. Les coupes immunohistologiques révèlent la présence d’une activité SA-β galactosidase au niveau de la tumeur préalablement traitée. A l’inverse, un autre patient ayant seulement
subi l’intervention chirurgicale ne présente pas d’activité SA-β galactosidase. De plus, chez ces deux patients, ce marqueur est peu présent voire indétectable au niveau des parties saines du poumon39. Les mêmes résultats
sont observés dans une autre étude portant sur une cohorte de patients souffrants d’un mésothéliome pleural malin et traité soit par trois cycles de Carboplatine et taxol ou bien 3 cycles de Cisplatine et Pemetrexed. La présence de marqueurs sénescents comme l’expression de p21 et une activité SA-β galactosidase est détectée an niveau de certaines tumeurs traitées. Ils sont significativement augmentés suivant le traitem ent en comparaison à la tumeur du même patient avant la thérapie néo-adjuvante 40. Également, dans le cas de tumeurs
liquides, la chimiothérapie induit l’entrée en sénescence des cellules tumorales. Effectivement, dans le modèle de lymphome murin EµMyc, Schmitt et al, montrent qu’un traitement avec l’agent alkylant, le Cyclophosphamide promeut l’établissement d’un état sénescent des cellules cancéreuses. Celles-ci ont une activité SA-β galactosidase, n’incorporent plus de BRDU et n’expriment pas l’antigène prolifératif Ki67 mais expriment à l’inverse p53. L’entrée en sénescence est dépendante de l’expression de p53 et p16. Les souris capables d’activer ce mécanisme suppressif présentent un meilleur pronostic que les souris déficientes pour l’expression de ces protéines 41.
Ainsi, la sénescence est également un mécanisme de suppression tumorale limitant la propagation de cellules tumorales suivant un traitement génotoxique.
2) Régulation du choix apoptose-sénescence suivant un traitement génotoxique 2a Le niveau de dommages de l’ADN module le choix apoptose-sénescence :
En réponse aux traitements génotoxiques, la sénescence n’est pas le seul mécanisme de suppression tumorale induit. En effet, l’apoptose se met également en place en réponse à la chimiothérapie in vitro et in
vivo 40,42. Ainsi, se pose la question du choix entre ces deux mécanismes de suppression tumorale. Une
première constatation est que le niveau de dommages de l’ADN semble devoir être plus élevé pour induire l’apoptose que la sénescence 43. Effectivement, le traitement de cellules tumorales mammaires MCF7 à
l’Adriamycine induit leur entrée en sénescence lorsqu’elles sont traitées à la concentration de 0,25µM alors qu’elles entrent en apoptose à la dose de 2µM 44. Également, le traitement de fibroblastes humains primaires
à de faibles concentrations d’Etoposide promeut une entrée en sénescence alors qu’à de plus fortes concentrations c’est l’apoptose qui est induite 45. L’activation de la voie des dommages de l’ADN conduit à
l’expression de p53 qui est nécessaire aussi bien au processus apoptotique qu’à la sénescence. La quantité de celle-ci semble également liée au choix du mécanisme de suppression tumorale. Effectivement, le traitement de fibroblastes humains diploïdes à l’H2O2 induit à la fois une entrée en sénescence et la mort. Les cellules en
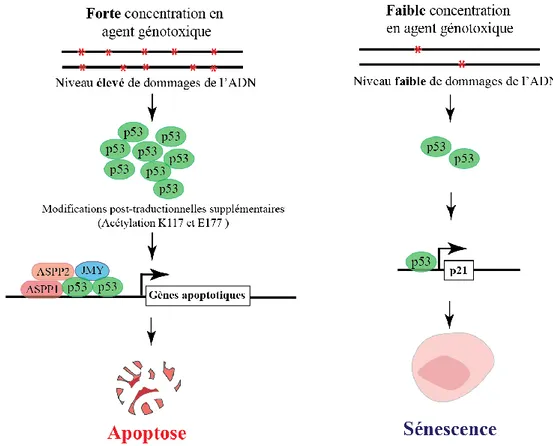
que p53 semble plus facilement induire p21 que les gènes pro-apoptotiques. En effet, la capacité de fixation de p53 sur le gène de p21 est plus importante que celle sur les gènes pro apoptotiques Bax et AIP1 47. Par
ailleurs, l’induction de gènes pro-apoptotiques requiert celle de co-facteurs transcriptionnels additionnels tels que JMY, ASPP1 ou ASPP2 dont l’expression n’est pas nécessaire à l’entrée en sénescence 48–50. De plus,
l’expression des gènes pro-apoptotiques par p53 requiert plusieurs modifications post-traductionnelles. Effectivement, la mutation K117R de p53 empêchant son acétylation sur ce site abolit l’entrée en apoptose suivant l’irradiation. Cependant, elle n’empêche pas l’induction de p21 et l’entrée en sénescence des cellules. De même, les souris arborant la mutation E177R de p53 perdent la capacité à entrer en apoptose alors que l’entrée en sénescence est maintenue 51.
Ainsi, l’induction de p21 par p53 semble plus facile que celles des gènes apoptotiques. En effet, l’expression de cet inhibiteur du cycle cellulaire par ce facteur de transcription requiert moins de modifications post-traductionnelles et de cofacteurs. Ceci peut expliquer pourquoi la sénescence nécessite généralement moins de dommage de l’ADN pour être induite que l’apoptose (Figure 4).
Figure 4 : La concentration en agent génotoxique et le niveau d’expression de p53 gouvernent le choix apoptose-sénescence.
Une concentration élevée en agent génotoxique est responsable d’une forte activation et stabilisation de p53, celle-ci permet la transcription des gènes apoptotiques et la mort des cellules. La sénescence requiert une concentration en agent génotoxique plus faible pour être induite. Ceci s’explique car p53 a une affinité plus forte pour le promoteur de p21 que les gènes apoptotiques et nécessite moins de cofacteurs transcriptionnels et de modifications post-traductionnelles pour favoriser l’expression de ce gène.
2b L’activation d’oncogènes comme AKT et la perte de suppresseurs de tumeur tels que PTEN modulent le choix apoptose-sénescence :
En plus du niveau de dommages, le choix entre l’apoptose et la sénescence est également gouverné par l’activation de certaines voies oncogéniques ou suppressives. Effectivement, une étude portant sur des lignées de gliome montre qu’en fonction du statut du suppresseur de tumeur PTEN, les cellules tumorales entrent en apoptose ou bien deviennent sénescentes. Les cellules déficientes entrent en sénescence suivant l’irradiation. A l’inverse, celles exprimant cette protéine meurent par apoptose. Dans les cellules PTEN déficientes, l’expression de la forme sauvage de cette protéine limite drastiquement l’entrée en sénescence suivant l’irradiation et favorise à l’inverse l’apoptose. PTEN étant un inhibiteur d’AKT, le rôle de cette kinase a également été étudié dans ce modèle. Sa déplétion dans les cellules déficientes pour l’expression de la protéine suppressive favorise leur entrée en apoptose indiquant qu’AKT bloque la mort cellulaire 52.
L’importance de cette kinase dans la régulation du choix apoptose-sénescence a également été appréhendée dans une étude du laboratoire. Effectivement, le traitement de lignées tumorales colorectales avec le métabolite actif de l’Irinotécan, le Sn38, induit durant les premières heures l’activation de cette kinase. Son inhibition pendant le traitement empêche leur entrée en sénescence et favorise l’apoptose 53. L’activation de cette kinase
semble donc nécessaire à la mise en place de ce mécanisme suppressif. Confirmant ces observations, les cellules primaires déficientes en AKT1 et 2 sont en partie résistantes à la sénescence réplicative ou l’OIS 54.
AKT favorise la mise en place du mécanisme suppressif en favorisant l’activation de la voie p53-p21 52,53,55.
Plus précisément, cette kinase promeut l’entrée en sénescence via l’activation de mTORC1. La voie AKT-mTORC1 induit la surexpression de p53 en augmentant sa traduction d’une part et en inhibant sa dégradation par Mdm2 d’autre part 55.
Ainsi, en fonction des voies oncogéniques et suppressives activées, les cellules tumorales entrent en sénescence ou bien en apoptose. L’entrée en sénescence semble privilégiée suivant la perte de certains suppresseurs de tumeurs comme PTEN ou bien la suractivation de voies oncogéniques telles qu’AKT-mTORC1. Par ailleurs, ce mécanisme suppressif requiert un niveau de dommage plus faible et que l’apoptose pour être induite. Ceci est expliqué par la plus forte capacité de fixation de p53 sur le promot eur de p21 que sur les gènes apoptotiques.
3) La dernière génération de thérapies anti-cancéreuses induit une entrée en sénescence
La sénescence n’est pas seulement induite par les thérapies génotoxiques. En effet, l’inhibiteur des kinases Cdk4/6, le Palbociclib, promeut la mise en place de ce mécanisme suppressif notamment dans des
agent dépend de p53 56. Cet effet est également retrouvé dans le cas du traitement de mélanomes résistants à
l’inhibiteur de B-Raf, le Vemurafenib. Ce résultat est également observé in vivo dans un modèle murin où des cellules de mélanome sensibles ou résistantes au Vemurafenib ont été greffées. Si le traitement au Pa lbociclib induit une régression tumorale dans les deux cas, seules les cellules résistantes au Vemurafenib entrent en sénescence 57. Par ailleurs, l’utilisation simultanée du Palbociclib et d’inhibiteurs de l’autophagie est
responsable de l’entrée en sénescence de lignées tumorales mammaires 58. Ainsi, l’inhibition ciblée de Cdk4/6
est responsable d’une entrée en sénescence de cellules tumorales in vitro et in vivo.
Par ailleurs, la sénescence se met également en place dans le cadre des thérapies exploitant le phénomène de létalité synthétique. Par exemple, l’inhibition de PARP1 dans des cellules tumorales arborant des mutations inactivatrices de BRCA1 ou BRCA2 est responsable de leur mort 59,60. Cependant, la sénescence
peut également se mettre en place dans ce contexte. En effet, son inhibition par l’Olaparib dans des lignées tumorales ovariennes induit l’activation de ce mécanisme suppressif dans certaines cellules. Celles-ci sont caractérisées par une activité SA-β galactosidase et représentent entre 30 et 70% de la population cellulaire en fonction de la lignée étudiée. Elles expriment par ailleurs les autres marqueurs typiques de l’état sénescent tels que l’expression d’inhibiteurs de Cycline/Cdk 61. De même, le traitement de lignées leucémiques avec des
inhibiteurs de PARP1 induit leur entrée en sénescence 62.
Ainsi, la sénescence comme l’apoptose est un mécanisme de suppression tumorale majeur induit in
vitro et in vivo après un traitement génotoxique, l’inhibition spécifique de protéines prolifératives telles que
Cdk4/6 ou bien exploitant le phénomène de létalité synthétique.
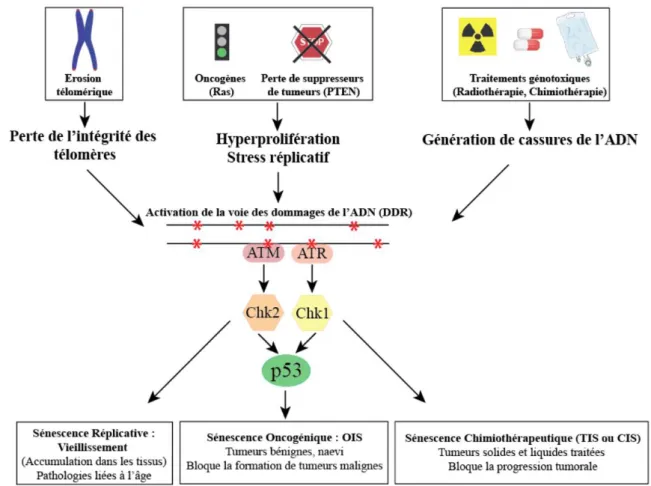
La sénescence constitue donc un mécanisme pléiotropique intervenant dans le vieillissement cellulaire, dans certaines pathologies, lors du développement embryonnaire, suite à l’activation d’un oncogène ou bien en réponse aux thérapies anti-cancéreuses. Elle constitue un mécanisme de suppression tumorale effectif stoppant la transformation cellulaire ou la prolifération des cellules tumorales lorsqu’elle est induite par la chimiothérapie (Figure 5).
Figure 5 : Mécanismes induisant la sénescence réplicative, oncogénique et chimiothérapeutique.
L’érosion télomérique au cours des divisions successives, l’hyperprolifération et les traitements génotoxiques génèrent des dommages de l’ADN. Les protéines de la voie des dommages de l’ADN sont nécessaires à l’établissement des trois types de sénescence. La sénescence réplicative est associée au vieillissement des cellules et participe au développement de pathologies liées à l’âge. La sénescence oncogénique constitue une barrière limitant la transformation. Ce type de sénescence est retrouvé au niveau de tumeurs bénignes comme les nævi. La sénescence chimiothérapeutique bloque la prolifération des cellules tumorales et est retrouvée au niveau de tumeurs malignes traitées.
L’entrée en sénescence repose sur l’expression et l’activité de plusieurs régulateurs qui entrainent de nombreuses modifications. La partie suivante est consacrée aux caractéristiques cellulaires et moléculaires des cellules sénescentes. Leur étude permet d’appréhender les profonds changements associés à l’établissement de ce mécanisme suppressif et d’identifier les cellules sénescentes.
II. Caractéristiques cellulaires et moléculaires des cellules sénescentes
A. Arrêt du cycle cellulaire :
1) Régulation de l’expression des inhibiteurs du cycle cellulaire p21 et p16.
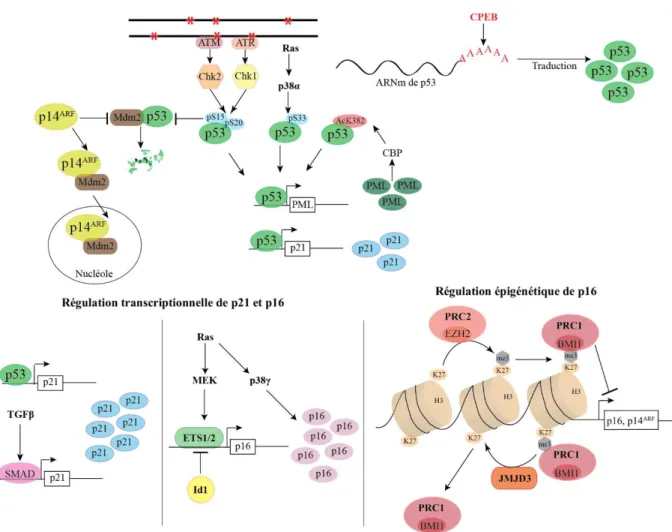
D’une manière générale, l’entrée en sénescence est la conséquence de dommages de l’ADN. L’induction de cette voie est responsable de l’activation d’ATR et ATM. Ces kinases activent ensuite Chk1 et Chk2 respectivement. En conséquence, p53 est phosphorylé sur la sérine 15 et 20. Ces modifications post-traductionnelles l’empêchent d’interagir avec l’E3 ubiquitine ligase Mdm2 responsable de sa dégradation. Ceci permet l’accumulation de p53 et augmente sa capacité à activer la transcription de ses gènes cibles 63–65.
Les acteurs clé de cette voie sont alors indispensables à l’entrée en sénescence 10,28,31,33. L’une des cibles de
p53, PML promeut son acétylation sur la lysine 382 par l’acétyltransférase CBP suivant la surexpression de l’oncogène Ras. Cette modification post-traductionnelle augmente son activité transcriptionnelle et favorise ainsi l’établissement de la sénescence 66,67. L’expression de p53 est également régulée par la protéine de
polyadénylation CPEB. Elle favorise la traduction de p53 par la polyadénylation de son ARN messager. L’expression de cette protéine est indispensable à l’entrée en sénescence réplicative de fibroblastes humains diploïdes 68. P53 une fois accumulée et activée induit l’expression de p21 69,70. Ces deux protéines sont
essentielles à l’activation du processus sénescent. Un défaut d’expression de l’une d’elles est responsable d’un contournement de l’état sénescent ou bien d’une entrée privilégiée en apoptose 53,71–74. La voie p53-p21
constitue dès lors une voie suppressive majeure permettant la mise en place de la sénescence. L’expression de p21, bien que majoritairement régulée par p53 peut également être induite indépendamment de celui-ci. En effet, dans le cadre de la sénescence oncogénique médiée par H-RasV12 de cellules épithéliales mammaires
humaines, l’expression de p21 requiert celle du récepteur de type II du TGFβ 75. Par ailleurs, elle peut être
régulée directement par Chk2. Dès lors, la simple surexpression de cette kinase est suffisante à induire une entrée en sénescence de cellules tumorales pulmonaires 76.
Une caractéristique fondamentale de ce mécanisme de suppression tumorale est l’arrêt prolifératif et sa persistance malgré la présence de signaux mitogéniques. En plus de p21, l’arrêt prolifératif requiert l’action de p16. L’expression de cet inhibiteur du cycle cellulaire est promue par ETS1 et ETS2 dont l’activité est induite par la kinase MEK. Ces deux facteurs de transcription se fixent sur le promoteur de p16 et favorisent ainsi sa transcription. L’inhibition de MEK dans les fibroblastes surexprimant Ras empêche l’induction de p16. A l’inverse, la protéine Id1 réprime transcriptionnellement l’expression de cet inhibiteur du cycle cellulaire. Ainsi, les fibroblastes en sénescence réplicative surexpriment ETS1 et ont une expression réduite
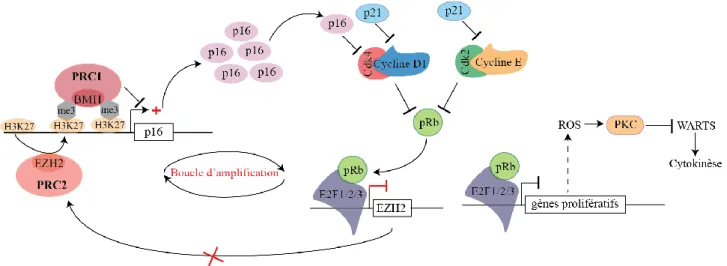
d’Id1 77. P16 est également finement régulée au niveau épigénétique par la protéine du complexe répresseur 2
des Polycomb (PRC2), EZH2. Celle-ci réprime transcriptionnellement p16 par la tri-méthylation de la lysine 27 de l’Histone H3 (H3K27me3) de son promoteur. Cette marque épigénétique est nécessaire au recrutement de la protéine du complexe répresseur 1 des Polycomb (PRC1), BMI1 qui est responsable de la répression transcriptionnelle de p16 78,79. A l’inverse, l’histone déméthylase JMJD3 retire la marque répressive générée
par EZH2 ce qui permet d’augmenter l’expression de cet inhibiteur du cycle cellulaire. Par ailleurs, en promouvant la déméthylation du locus INK4A-ARF, JMJD3 favorise également l’expression de p14ARF située
sur le même locus que p16 80. Cette protéine suppressive promeut la stabilisation de p53 en empêchant sa dégradation par Mdm2. Plus précisément, p14ARF interagit avec Mdm2 et induit sa séquestration nucléolaire 81–86. Dans les fibroblastes embryonnaires murins, l’inhibition de JMJD3 réduit l’expression de p16 et p19ARF
(p14ARF chez l’Homme)et induit ainsi leur immortalisation 80.
L’expression p16 et p21 est également promue par la kinase p38MAPK. Plus particulièrement, les isoformes p38α et p38γ sont activés suivant la surexpression de l’oncogène Ras. P38γ est nécessaire à l’induction de p16 dans ce contexte. Quant à lui, p38α active p53 en le phosphorylant sur la sérine 33 ce qui augmente l’expression de p21. Dès lors, la déplétion de p38α ou p38γ boque l’entrée en sénescence oncogénique 87.
Ainsi, l’entrée en sénescence s’accompagne de l’accumulation de dommages de l’ADN activant le facteur de transcription p53 responsable de l’expression de p21. Également, la protéine suppressive de tumeur p16 est induite. L’expression de p21 et p16 constitue une caractéristique des cellules sénescentes qui est mise à profit pour leur identification. Cependant, ces deux inhibiteurs du cycle cellulaire ne sont pas seulement des marqueurs l’état sénescent. Effectivement, elles sont nécessaires à la mise en place et au maintien de ce mécanisme suppressif (Figure 6).