© Raphael Lafleur Lambert, 2018
Études de modificateurs pour le transfert de chiralité
sur une surface de platine & Synthèse totale de
l'(+)-O-méthylasparvenone et d'autres métabolites fongiques
avec intérêt biothérapeutique
Thèse
Raphael Lafleur Lambert
Doctorat en chimie
Philosophiæ doctor (Ph. D.)
Études de modificateurs pour le transfert de
chiralité sur une surface de platine
&
Synthèse totale de l’(+)-O-méthylasparvenone et
d’autres métabolites fongiques avec intérêt
biothérapeutique
Thèse
Raphaël Lafleur-Lambert
Sous la direction de :
John Boukouvalas, directeur de recherche
Peter McBreen, codirecteur de recherche
iii
Résumé
Les travaux de cette thèse sont, en partie, les fruits d’une codirection entre le laboratoire des surfaces dirigé par le professeur Peter McBreen et le laboratoire de synthèse organique dirigé par le professeur John Boukouvalas. Toutes les images ont été obtenues par les étudiants au laboratoire de surfaces et mes travaux consistaient à participer aux synthèses des composés organiques. L’autre partie concerne des projets en synthèse totale.
Dans le premier chapitre, il y a la présentation de la réaction d’Orito, une réaction catalytique hétérogène asymétrique, utilisant des alcaloïdes de la Cinchona et des dérivés analogues comme modificateur chiral.
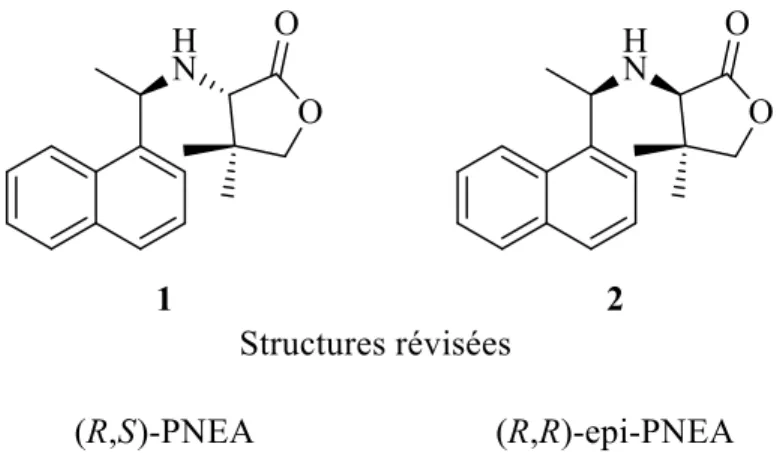
Le chapitre II, à la partie A, concerne la compréhension des structures de modificateurs chiraux et de leur rôle associés au transfert de chiralité au cours de la réaction d’Orito. Les méthodes de chimie organique, soit l’hydrogénolyse du composé suivi de la synthèse d’un dérivé connu, ont permis de prouver que la structure absolue du modificateur synthétique de grande importance PNEA est R, S. L’importance de cette correction vient au fait que la structure du composé est directement liée à sa performance, en comparant avec son épimère qui ne possède pas les mêmes activités de transfert de chiralité. Ces travaux ont été publiés dans le journal ACS Catalysis. Au chapitre II partie B, c’est l’étude du transfert de chiralité d’un modificateur synthétique unique ne portant pas de groupement azote que nous avons synthétisé pour le laboratoire des surfaces avec la dihydroxylation de Sharpless. L’absence de fonction azote est très révélatrice quant à la compréhension mécanistique du transfert de chiralité. Cette absence démontre que le design de modificateur ne nécessite pas ce type d’atome dans sa structure. On retrouve l’article sur ce sujet dans le journal Surface
Science.
Le chapitre III contient la première synthèse d’un produit naturel, l’antrocinnamomin D, membre d’une famille de composés issu d’un champignon appelé Niuchangchih. La méthode de synthèse employée permet aussi d’obtenir deux autres membres de la famille, soit les antrodins A et B, un anhydride maléique et un maléimide. L’antrocinnamomin D,
iv
l’antrodin A et B ont été obtenus successivement en 6-8 étapes avec des rendements de 51%, 46% et 43% respectivement. Cette synthèse inclut un couplage croisé qui s’est démontré très efficace. Ce couplage fait intervenir comme catalyseur métallique un composé à base de Fe, un élément abondant et peu toxique, qui a su rivaliser le Pd, un des catalyseurs de métaux nobles habituels. Cette synthèse se retrouve dans le journal Tetrahedron.
Le chapitre IV porte sur la synthèse totale d’un produit naturel rare sans azote actif comme antagoniste de la sérotonine. Ce produit a été isolé d’un champignon de sol appelé
Aspergillus parvulus. Son rôle antagoniste se situe au récepteur 2C de la sérotonine. Ce
récepteur est associé aux troubles liés à la dépression et les agents antagonistes ont démontré des effets bénéfiques pour les patients atteints de ce problème de santé. Cette antagoniste naturelle appelée (+)-O-méthylasparvenone fait partie d’une famille de composés naturels appelée 4-hydroxy-1-tetralone. Leur structure simpliste cache une complexité exprimée par la rareté de synthèse énantiosélective de ces composés. La synthèse asymétrique de l’antagoniste, une 4-hydroxy-1-tetralone trisubstituée, a été accomplie avec un excès d’énantiomère de 94% en 8 étapes avec un rendement global de 22%. Cette synthèse inclut trois étapes clés, dont une alkylation réductrice, une alkynylation asymétrique et une acylation de Friedel-Crafts. Cette synthèse a été rapportée dans le journal Organic &
Biomolecular Chemistry.
Au chapitre V, on y retrouve une synthèse totale unie et régiosélective de deux composés de la famille des rubrolides, soit les membres R et S. Ces deux composés sont issus du champignon Aspergillus terreus OUCMDZ-1925. Le rubrolide R a été décrit comme un antioxydant et le rubrolide S a démontré des activités anti-influenza A(H1N1). Les deux approches de synthèse font intervenir à la fois une condensation de type aldol vinylogue et un couplage de Suzuki comme étapes clés. Ce projet donne accès au rubrolide R en 6 étapes par une voie de synthèse avec un rendement de 8,9% et au rubrolide S avec une étape supplémentaire pour un rendement global de 8,5%.
v
Summary
The work of this thesis is, in part, the result of a co-direction between the surface laboratory headed by Professor Peter McBreen and the organic synthesis laboratory led by Professor John Boukouvalas. All the images were obtained by the students in the surface laboratory and my work consisted of taking part in syntheses of the organic compounds. The other part concerns projects in total synthesis.
In the first chapter, there is the presentation of the Orito reaction, an asymmetric heterogeneous catalytic reaction, using Cinchona alkaloids and analogous derivatives as a chiral modifier.
Chapter II, Part A, discusses the understanding of chiral modifier structures and their role associated with the chiral transfer during the Orito reaction. The methods of organic chemistry, namely the hydrogenolysis of the compound followed by the synthesis of a known derivative, have made it possible to prove that the absolute structure of the synthetic modifier PNEA is R, S. The importance of this correction comes from the fact that the structure of the compound is directly related to its performance, comparing with its epimer which does not have the same chiral transfer activities. These works were published in the journal ACS
Catalysis. In Chapter II, Part B, it is the study of the transfer of chirality of a single synthetic
modifier not carrying a nitrogen group that we have synthesized for the laboratory of the surfaces with the dihydroxylation of Sharpless. The absence of nitrogen function is very revealing as to the mechanistic understanding of chirality transfer. This absence demonstrates that the modifier design does not require this type of atom in its structure. The article on this subject can be found in the journal Surface Science.
Chapter III contains the first synthesis of a natural product, antrocinnamomin D, a member of a family of compounds derived from a fungus called Niuchangchih. The method of synthesis used also makes it possible to obtain two other members of the family, the antrodins A and B, a maleic anhydride and a maleimide. Antrocinnamomin D, antrodin A and B were successively obtained in 6-8 steps with yields of 51%, 46% and 43% respectively.
vi
This synthesis includes a cross-coupling that has been shown to be very effective. This coupling involves as metal catalyst a compound based on Fe, an abundant and low toxicity element, which has been able to compete with Pd, one of the usual noble metal catalysts. This synthesis is found in the journal Tetrahedron.
Chapter IV deals with the total synthesis of a rare natural product without nitrogen active as a serotonin antagonist. This product was isolated from a soil fungus called
Aspergillus parvulus. Its antagonistic role is at the 2C receptor of serotonin. This receptor is
associated with depression-related disorders and the antagonistic agents have shown beneficial effects for patients with this health problem. This natural antagonist called (+)-O-methylasparvenone is part of a family of natural compounds called 4-hydroxy-1-tetralone. Their simplistic structure hides a complexity expressed by the scarcity of enantioselective synthesis of these compounds. The asymmetric synthesis of the antagonist, a trisubstituted 4-hydroxy-1-tetralone, was accomplished with an enantiomeric excess of 94% in 8 steps with an overall yield of 22%. This synthesis includes three key steps, including reductive alkylation, asymmetric alkynylation, and Friedel-Crafts acylation. This synthesis was reported in the journal Organic & Biomolecular Chemistry.
In Chapter V, we find a total, unified and regioselective synthesis of two compounds of the rubrolide family, the R and S members. These two compounds are derived from the fungus Aspergillus terreus OUCMDZ-1925. Rubrolide R has been reported as an antioxidant and rubrolide S has been shown to have anti-influenza A (H1N1) activity. Both synthesis approaches involve both a vinylogous aldol condensation and a Suzuki coupling as key steps. This project gives access to rubrolide R in 6 steps by a synthetic route with a yield of 8.9% and to rubrolide S with an additional step for an overall yield of 8.5%.
vii
Table des matières
Résumé ...iii
Summary ... v
Liste des abréviations ... xvi
Remerciements ... xxi
Chapitre I : Introduction ... 1
La chiralité ... 2
La catalyse ... 4
La catalyse homogène ... 5
La catalyse homogène métallique ... 5
La catalyse homogène asymétrique ... 7
La biocatalyse ... 8
La catalyse hétérogène ... 10
La catalyse hétérogène asymétrique ... 11
La catalyse au Ni ... 12
La catalyse au Pd ... 13
La catalyse au Pt : la réaction d’Orito ... 13
STM... 21
Chapitre II partie A : Études de la réduction du cétopantolactone avec la réaction d’Orito ... 26
Introduction ... 27
Travaux présentés ... 32
Conclusions ... 46
Chapitre II partie B : Études spectroscopiques du NED et deux analogues ... 48
Introduction ... 49 Travaux présentés ... 50 NEA ... 52 NED ... 57 MNEA ... 60 Conclusions ... 64
Chapitre III : Première synthèse de l’antrocinnamomin D ... 65
Introduction ... 66
Travaux présentés ... 80
viii
Synthèse ... 82
Conclusions ... 85
Chapitre IV : Synthèse de l’(+)-O-méthylasparvenone ... 86
Introduction ... 87
Synthèse énantiosélective de la famille 4-hydroxy-tetralone ... 91
Zéro substituant ... 91
Un substituant ... 94
Deux substituants et plus ... 94
Synthèse de l’(±)-O-méthylasparvenone ... 94
Travaux présentés ... 99
Rétrosynthèse ... 99
Tentatives de synthèse du (S)-asparvenone ... 114
Conclusions ... 115
Chapitre V : Synthèse unifiée régiosélective des rubrolides R et S ... 117
Introduction ... 118
Les composés antiviraux ... 119
Historique sur les rubrolides ... 120
Première synthèse des rubrolides R et S ... 122
Travaux présentés ... 126
Rétrosynthèse ... 126
Synthèse du rubrolide R ... 132
Conclusions ... 138
Chapitre VI : Conclusions générales ... 140
Partie expérimentale ... 144
ix
Liste des figures
Figure 1 : Présentation de la distinction des objets symétriques et des objets chiraux. Les objets achiraux sont superposables avec leur image miroir, mais pas les objets chiraux. ... 2 Figure 2 : Importance du contrôle de la chiralité dans les médicaments. L’exemple de l’éthambutol montre qu’un de ses énantiomères est un remède et l’autre est un composé qui provoque la cécité. . 3 Figure 3 : Coordonnée de réaction pour la conversion du produit X en Y. Réaction non catalysée (noir). Réaction catalysée (rouge). ... 4 Figure 4 : Quatre pseudo-énantiomères extraits de la Cinchona. Les deux centres chiraux
représentés distinguent les pairs CD/CN et QN/QD. Le groupement X (H, OMe) distingue les composés CN/CD des composés QN/QD. L’amine chiral bicyclique appelée quinuclidine est la même pour les quatre composés, empêchant ainsi les composés d’être de vrais pairs
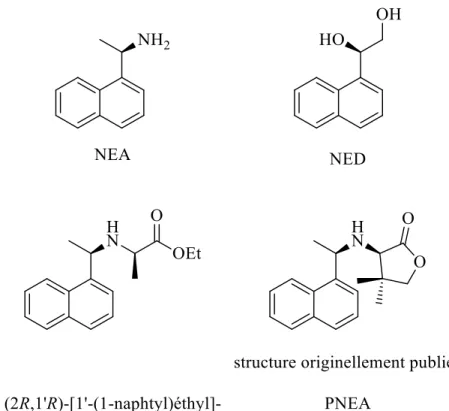
d’énantiomères. ... 15 Figure 5 : Ligand chiral du mélange AD-mix bêta composé de deux molécules de dihydroquinidine et d’une molécule de phtalazine (représenté en bleu). ... 17 Figure 6 : Quatre modificateurs chiraux synthétiques. Le NEA est le seul modificateur commercial. Le (2R,1'R)-[1'-(1-naphtyl)éthyl]-2-amino propionoate d'éthyle et le PNEA avec sa structure
originellement publiée sont obtenus à partir du NEA. ... 19 Figure 7 : Trois coupures de cristaux selon l’indice de Miller. Les coordonnées en parenthèse représentent successivement l’interaction du plan avec l’axe des abscisses, l’axe des ordonnées et l’axe des cotes. ... 20 Figure 8 : Jonction tunnel entre une pointe de W et la surface du platine. L’axe des abscisses
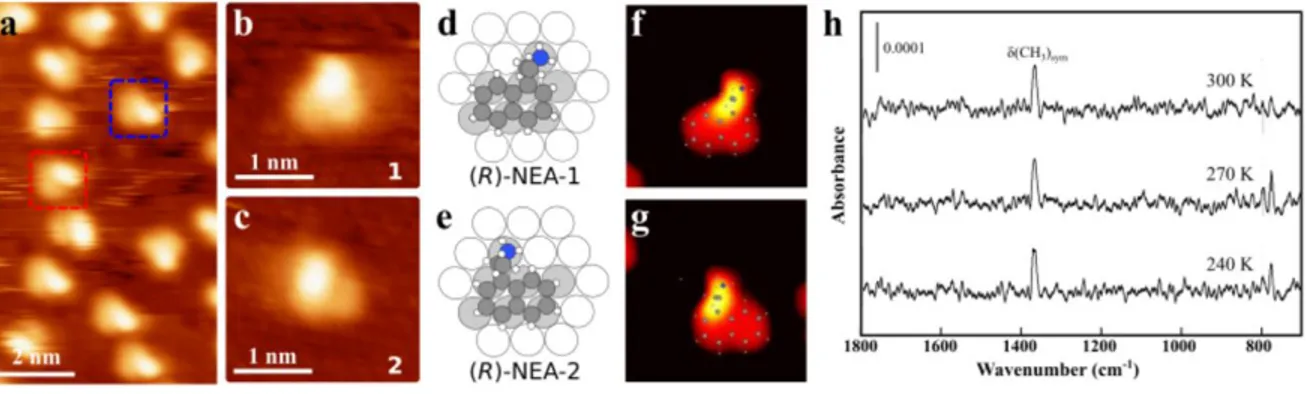
représente la distance pointe-surface et l’axe des ordonnées le courant. ... 22 Figure 9 : (a) Appareillage du microscope à effet tunnel (model : Specs Aarhus 150 STM). (b) Le scanneur y compris le tuyau piézoélectrique (5 et 9), la pointe (4) et l’échantillon (1). ... 23 Figure 10 : Détermination de la géométrie d'adsorption du (S)-NEA, sur une image STM Pt(111). L’image adaptée (a) montre plusieurs molécules de (R)-NEA adsorbées sur Pt(111). Les carrés rouges et bleus mettent en évidence l'observation de deux motifs STM distincts. Les deux motifs, présentés séparément dans (b, c), sont étiquetés 1 et 2, respectivement. Conditions de tunneling: Vt = 1 V; il = 0,200 nA. Les images (d, e) représentent les deux géométries de chimisorption les plus stables calculées par DFT. L'atome N est représenté en bleu et les atomes de Pt auxquels le (R)-NEA est directement coordonné sont ombrés. Les images (f, g) sont les motifs STM simulés par DFT pour les structures (d, e), respectivement. L’image (h) représente les spectres RAIRS du (R)-NEA sur Pt(111) dans la gamme de températures explorée dans les mesures STM. ... 24 Figure 11 : Structures originellement publiées du PNEA et de son épimère par le groupe de Baiker et al. à partir d’études NOE. Lorsque le méthyle du premier centre chiral est irradié, le signal de l’hydrogène du deuxième centre chiral est augmenté. Ensuite, le signal de l’hydrogène du premier centre chiral est augmenté lorsque le signal méthyle pointant vers le bas de la lactone est augmenté. ... 31 Figure 12 : Structures révisées du PNEA avec les centres chiraux (R,S) par les groupes de
Boukouvalas et McBreen de l’Université Laval. ... 32 Figure 13 : Rationalisation de l’image STM du PNEA. La structure moléculaire du PNEA est divisible en trois parties distinctes représentées par le groupement lactone (1), le groupement éthyle
x
(2) et le groupement naphtyle (3). Par souci de clarté, l’image STM est incluse une seconde fois sans les numéros. ... 33 Figure 14 : Images STM sur Pt(111) à 300 K. (A-D) adsorption du PNEA. (E) représente un
fragment issu du PNEA. (F) représente le fragment vinylnaphtalène déjà étudié par STM. (Goubert, G.; Demers-Carpentier, V.; Masini, F.; Dong, Y.; Lemay, J. C.; McBreen, P. H. Chem. Commun. 2011, 47, 9113) ... 34 Figure 15 : Images STM attribuées au fragment AF adsorbé sur le Pt(111) à 300 K :(A) monomère, (B) dimère et (C) trimère. ... 36 Figure 16 : Images STM d’assemblages supramoléculaires par la co-adsorption du AF et du KPL sur le Pt(111) à 300 K. ... 41 Figure 17 : Images STM d’assemblages supramoléculaires par la co-adsorption du PNEA, AF et du KPL sur le Pt(111) à 300 K. Les assemblages A-E sont attribués à des structures impliquant le AF formé lors de l’hydrogénolyse du PNEA sur la surface de Pt(111). ... 42 Figure 18 : Images STM de l’évolution sur le temps (0, 80, 160, 240 et 520s) des assemblages KPL et AF sur le Pt(111) à 300 K. ... 43 Figure 19 : (A) Image STM d’un balayage élargi de AF avec TFAP sur le Pt(111) à 300 K. (B-C) Images STM de complexes isolés de AF avec le TFAP sur le Pt(111) à 300 K. ... 45 Figure 20 : Images STM du (R)-NEA sur le Pt(111) à 300 K et représentation schématique de la géométrie du (R)-NEA-1 et 2. ... 52 Figure 21 : Images STM des complexes NEA/KPL sur le Pt(111). Les motifs (A) et (B) impliquent le (R)-NEA-1 et les motifs (C-D-E) le (R)-NEA-2. Des propositions pour les structures moléculaires correspondantes aux images sont montrées en dessous des images. ... 54 Figure 22 : Représentation schématique de la distribution en structure et en prochiralité du KPL dans les complexes KPL/NEA sur le Pt(111). ... 56 Figure 23 : Images STM du (R)-NED sur le Pt(111) à 300 K et représentation schématique de la géométrie du (R)-NED-1 et 2. ... 57 Figure 24 : Images STM des complexes NED/KPL sur le Pt(111) à 300 K. Les motifs (A) et (B) impliquent le (R)-NED-1 et les motifs (C-D) le (R)-NED-2. Des propositions pour les structures moléculaires correspondantes aux images sont montrées en dessous des images. ... 58 Figure 25 : Représentation schématique de la distribution en structure et en prochiralité des
complexes KPL/NED observés par STM sur le Pt(111). ... 59 Figure 26 : Images STM du (R)-MNEA sur le Pt(111) à 300 K et représentation schématique de la géométrie du (R)-MNEA-1 et 2. ... 60 Figure 27 : Images STM des complexes MNEA/KPL sur le Pt(111) à 300 K. Les motifs (A) et (B) impliquent le (R)-MNEA-1 et les motifs (C) le (R)-MNEA-2. Des propositions pour les structures moléculaires correspondantes aux images sont montrées en dessous des images. ... 61 Figure 28 : Représentation schématique de la distribution en structure et en prochiralité des
complexes KPL/MNEA observés par STM sur le Pt(111). ... 62 Figure 29 : Antrodins A, B et leur analogue naturel l’antrocinnamomin D. ... 70 Figure 30 : Le neurotransmetteur sérotonine (5-HT), son antagoniste (20) (+)-O-méthylasparvenone et la structure de la famille des 4-hydroxy-1-tetralone. ... 87 Figure 31 : Six produits naturels membres de la famille des 4-hydroxy-1-tetralone. Les trois
xi
Figure 32 : Représentation d’une synapse avec les composantes présentes entre deux neurones. Le mode d’action des médicaments ISRS sur les SERT sont représentés par l’emplacement des figures géométriques. On observe aussi l’interaction de l’antagoniste 5-HT2C dans le récepteur 5-HT2C. ... 89
xii
Liste des schémas
Schéma 1 : Couplage croisé par catalyse homogène au fer lors de la synthèse de l’Antrocinnamomin D du chapitre III. La liaison C-C est formée entre le buténolide et le réactif de Grignard par la présence catalytique de l’acétylacétonate de Fe (III). ... 6 Schéma 2 : Couplage catalysé au palladium utilisé lors de la synthèse du rubrolide S. La formation du lien C-C entre le bromobuténolide et l’acide boronique est catalysée par le Pd(PPh3)2Cl2. ... 7
Schéma 3 : Oxydation régiosélective par catalyse au Pd encombré par le ligand naturel (-)-spartéine lors de la synthèse énantiosélective d’une 4-hydroxy-1-tetralone à partir d’un diol méso. ... 8 Schéma 4 : Réduction énantiosélective par catalyse enzymatique d’un composé dione en 4-hydroxy-1-tetralone. ... 9 Schéma 5 : Réduction par catalyse hétérogène d’un alcyne en alcane par hydrogénation sur Pd/C en présence d’hydrogène gazeux (50 psi). ... 10 Schéma 6 : Hydrogénation catalytique au Ni modifié par l’acide tartrique de cétones
fonctionnalisées en bêta. ... 12 Schéma 7 : Hydrogénation énantiosélective de cétones fonctionnalisées en alpha à l’aide de Pt en présence d’un modificateur chiral, communément appelée la réaction d’Orito. ... 13 Schéma 8 : Hydrogénation énantiosélective publiée par Orito. Conversion du benzoylformate d’éthyle en mandelate d’éthyle sur le Pt modifié par la cinchonidine. ... 14 Schéma 9 : Synthèse du NED par dihydroxylation à partir du vinylnaphtalène avec le mélange Ad-mix bêta. ... 16 Schéma 10 : Réarrangement de Kornblum-DeLaMare asymétrique sur un composé diène avec l’utilisation d’oxygène singulet et de quinidine comme auxiliaire chirale. ... 18 Schéma 11 : Réduction énantiosélective du cétopantolactone (KPL) en pantolactone (PL) en
fonction de deux diastéréoisomères agissant comme modificateur chiral. La différence du centre chiral entre le PNEA (1) et de l’epi-PNEA (2) génère une différence de 34% en excès
énantiomérique. ... 27 Schéma 12 : Synthèse de la vitamine B5 à partir de l’alcool chiral pantolactone qu’on peut obtenir à partir de l’hydrogénation asymétrique du KPL. ... 28 Schéma 13 : Hydrogénation énantiosélective du KPL sur Pt avec la CD comme modificateur chiral selon deux procédés de synthèse. La première est par traitement par lots avec un réacteur discontinu offrant un excès énantiomérique de 92% et l’autre procédé est par production en continu offrant un excès énantiomérique de 83%. ... 29 Schéma 14 : Hydrogénation énantiosélective avec le modificateur chiral PNEA dans le toluène en présence d’acide trifluoroacétique (TFA) offrant un excès énantiomérique de 79%. ... 30 Schéma 15 : Synthèse par amination réductrice du PNEA et de son épimère à partir du
cétopantolactone et du (R)-NEA dans le Ti(O-i-Pr)4 en présence de NaBH3CN. ... 30
Schéma 16 : Hydrogénolyse du PNEA sur le Pt à température pièce sous ultra haut vide (UHV). .. 35 Schéma 17 : Dégradation du PNEA à 300 K sous UHV amenant au fragment vinylnaphtalène. .... 35 Schéma 18 : Synthèse du fragment AF à partir du PNEA par hydrogénolyse sur Pd en présence d’hydrogène gazeux. ... 37
xiii
Schéma 19 : Synthèse d’un dérivé BOC déjà connu et caractérisé dans la littérature à partir du
composé AF. ... 37
Schéma 20 : Synthèse et caractérisation avec le pouvoir rotatoire du (S)-(+)-BOC-AF tel que publié dans la littérature par Freskos. ... 38
Schéma 21 : Synthèse du AF dans les conditions réactionnelles de l’hydrogénation asymétrique hétérogène. ... 39
Schéma 22 : Image STM de l’assemblage formé par la coadsorption du AF et du KPL sur le Pt(111) à 300K. ... 41
Schéma 23 : Énantiosélectivité de l’hydrogénation du KPL sur le Pt avec différents modificateurs chiraux. ... 49
Schéma 24 : Synthèse des Antrodins A et B par le groupe de Argade. ... 72
Schéma 25 : Synthèse des Antrodins A et B par Stewart et ses collègues. ... 74
Schéma 26 : Synthèse des antrodins A et B par Lee et ses collègues. ... 76
Schéma 27 : Les trois dernières étapes de la synthèse totale de l’epiccocin G par le groupe de Nicolaou dont la première étape fait intervenir de l’oxygène singulet menant au réarrangement de Kornblum-DeLaMare. ... 78
Schéma 28 : Synthèse du sandresolide B par le groupe de Trauner utilisant un réarrangement de Kornblum-DeLaMare. ... 79
Schéma 29 : Synthèse par le groupe de Clive du mélange contenant le produit naturel microperfuranone. ... 80
Schéma 30 : Rétrosynthèse de l’antrocinnamomin D et les antrodins A et B. ... 81
Schéma 31 : Préparation du diméthyldioxirane (DMDO) à partir d’acétone réagissant avec l’oxone. ... 82
Schéma 32 : Première synthèse de l’antrocinnamomin D. ... 84
Schéma 33 : Synthèse d’un 4-hydroxy-1-tetralone asymétrique avec le catalyseur CBS à partir d’une dicétone. ... 92
Schéma 34 : Synthèse asymétrique d’un 4-hydroxy-1-tetralone à partir d’un diol racémique par catalyse homogène et enzymatique. ... 93
Schéma 35 : Synthèse de l’O-méthylasparvenone racémique par le groupe de Brassard de l’Université Laval. ... 95
Schéma 36 : Synthèse de l’O-méthylasparvenone racémique par Bös. ... 96
Schéma 37 : Synthèse de l’intermédiaire clé A par Zard. ... 97
Schéma 38 : Synthèses de deux 4-hydroxy-1-tetralones racémiques par Zard, soit l’O-méthylasparvenone et le 10-norparvulenone et la synthèse possible du seimatorone racémique par leur approche. ... 98
Schéma 39 : Rétrosynthèse incluant les étapes clés soit l’acylation de Friedel-Crafts, l’alkynylation asymétrique et l’alkylation réductrice. ... 99
Schéma 40 : Mécanisme proposé de la synthèse de l’aldéhyde à l’aide de deux SET (transfert d’un seul électron). ... 101
Schéma 41 : Origine du produit commercial : l’acide tannique. ... 102
Schéma 42 : Approche préliminaire problématique de la synthèse de l’aldéhyde causé par la présence d’un produit secondaire. ... 103
xiv
Schéma 43 : Ajustement de la synthèse de l’aldéhyde par remplacement du réactif du bromoéthane
par l’iodoéthane. ... 104
Schéma 44 : Alkynylation asymétrique d’aldéhydes par la méthode de Pu. ... 104
Schéma 45 : Alkynylation asymétrique pour la synthèse du (-)-panacène auparavant employé par le groupe de Boukouvalas. ... 105
Schéma 46 : Alkynylation asymétrique d’aldéhydes par la méthode de Trost. ... 106
Schéma 47 : Conditions optimisées pour l’alkynylation asymétrique permettant d’obtenir l’alcool propargylique désiré. ... 108
Schéma 48 : Synthèse de l’acide intermédiaire clé 17 à partir de l’alcool 14. ... 109
Schéma 49 : Le choix du groupement protecteur en fonction des conditions de Friedel-Crafts employés pour la cyclisation a été le TBDPS. Ce groupement est plus résistant que le groupement TBDMS et ne génère pas le produit secondaire de lactonisation. ... 110
Schéma 50 : Cyclisation avec la réaction de Friedel-Crafts démontrant la symétrie caché du substrat. ... 111
Schéma 51 : Synthèse du produit naturel (+)-O-méthylasparvenone à partir de l’intermédiaire diol protégé par la fonction méthoxy et TBDPS. ... 111
Schéma 52 : Synthèse envisagée de l’(+)-asparvenone à partir d’un de nos intermédiaires (19). .. 114
Schéma 53 : Perspectives de synthèse à partir de notre méthode pour obtenir des 4-hydroxy-1-tetralone. ... 116
Schéma 54 : Synthèse du buténolide clé pour obtenir le rubrolide E à partir du bromolactone et de l’acide boronique. ... 120
Schéma 55 : Méthode précédente pour obtenir le rubrolide E du laboratoire Boukouvalas. ... 121
Schéma 56 : Les deux dernières étapes de synthèse du rubrolide E par le groupe d’Argade. La première est une condensation aldol vinylogue et la seconde est une déprotection des phénols. ... 121
Schéma 57 : Construction du buténolide clé par Jun à partir d’une acétophénone substituée. ... 123
Schéma 58 : Synthèse des aldéhydes A et B pour les rubrolides S et R par Jun. ... 124
Schéma 59 : Condensation et déprotection pour obtenir des rubrolides R et S par Jun. ... 125
Schéma 60 : Rétrosynthèse unie pour les rubrolides R et S avec les étapes clés de condensation aldol vinylogue en premier lieu et de couplage de Suzuki en second lieu. ... 126
Schéma 61 : Préparation du bromolactone. ... 127
Schéma 62 : Synthèses des aldéhydes pour les rubrolides R et S. ... 127
Schéma 63 : Préparation alternative de fonctions chromane retrouvées dans la littérature. ... 128
Schéma 64 : Tentative infructueuse de synthèse de l’aldéhyde chromane désiré. ... 128
Schéma 65 : Synthèse du buténolide par condensation aldol vinylogue. ... 129
Schéma 66 : Échec d’application de la réaction de condensation dans les conditions de Knoevenagel. ... 129
Schéma 67 : Synthèse du rubrolide S avec un rendement quantitatif par déprotection du produit de couplage. ... 132
Schéma 68 : Échec d’une synthèse alternative de l’aldéhyde par échange halogène-lithium. ... 133
Schéma 69 : Synthèse du buténolide par condensation aldol vinylogue à partir de l’aldéhyde. ... 133
Schéma 70 : Échec de la protection suivi de la condensation aldol vinylogue avec l’approche à un seul pot. ... 134 Schéma 71 : Déprotection avec le TBAF pour obtenir le rubrolide R avec un rendement de 90%. 137
xv
Liste des tableaux
Tableau 1 : Optimisation des différentes conditions pour l’alkynylation pour obtenir l’alcool asymétrique désiré. * La méthode de Trost a été utilisée dans ce cas. L’entrée 6 a fourni les
conditions optimales pour les besoins de la synthèse. ... 107 Tableau 2 : Comparaison de différentes données RMN de la littérature pour
l’O-méthylasparvenone. ... 113 Tableau 3 : Optimisation du couplage avec l’acide boronique méthylé. L’entrée 3 a offert les conditions pour obtenir le meilleur rendement. * Le signe % représente le rendement. ... 130 Tableau 4 : Optimisation du couplage avec l’acide boronique sans groupement protecteur. L’entrée 1 a été les conditions optimales pour le couplage. ... 131 Tableau 5 : Couplage de Suzuki avec l’acide boronique contenant le groupement TBS. L’entrée 2 a offert les meilleures conditions avec un rendement de 76%. ... 135 Tableau 6 : Couplage de Suzuki sans groupement protecteur pour le rubrolide R. La seule entrée pour ce couplage qui a offert du produit est l’entrée 1 avec un rendement de 39%. ... 136 Tableau 7 : Optimisation de la cyclisation pour convertir le rubrolide R en S. L’entrée 1 a permis de convertir le rubrolide R en S avec un rendement de 95%. ... 138
xvi
Liste des abréviations
# 5-HT 5-hydroxytryptamine (sérotonine) 9-BBN 9-borabicyclo[3.3.1]nonane A [α] pouvoir rotatoire Å angström Ac acétyle acac acétylacétonate AF amino-4,4-diméthyldihydrofuran-2-one aq aqueux atm atmosphère B BINOL 1,1′-bi-2-naphtol Bn benzyle Boc tert-butoxycarbonyle
br large (broad) spectre
n-Bu butyle s-Bu sec-butyle t-Bu tert-butyle Bz benzoyle C °C degrés Celsius calcd calculated cat catalytique cm-1 nombre d’onde CN cinchonine CD cinchonidine
COSY spectroscopie par corrélation D δ déplacement chimique d doublet dba dibenzylidèneacétone DBP Phtalate de dibutyle DBU 1,8-diazabicyclo[5.4.0]undec-7-ène DCM dichlorométhane DCE 1,2-dichloroéthane DMF diméthylformamide DMDO diméthyldioxirane DMSO diméthylsulfoxyde E ee excès d’énantiomère equiv équivalent Et éthyle
xvii F
G
GP groupe protecteur H
HMBC corrélation hétéronucléaire par multiples liens HMDS bis(triméthylsilyl)amine
HMPA triamide hexaméthylphosphorique
HMQC corrélation quantique multiple hétéonucléaire
HPLC chromatographie en phase liquide à haute performance HRMS spectrométrie de masse haute résolution
HSQC corrélation quantique unique hétéonucléaire hz hertz
I
IR infra rouge
ISRS inhibiteur sélectif de la recapture de la sérotonine J
J constante de couplage (spectre) K k kilo K kelvin KPL cétopantolactone L L litre
LDA diisopropylamide de lithium Lit. littérature
M
µ micro
m multiplet (spectre) Me méthyle
MNEA (R)-N-méthyle-1-(1-naphtyl) éthylamine mp melting point (point de fusion)
Ms mésylate m/z masse/charge N
ν fréquence (spectre)
NADP+ nicotinamide adénine dinucléotide phosphate NBS N-bromosuccinimide
NEA (R)-1-(1-naphtyl) éthylamine NED (R)-1-naphtyl-1,2-éthandediol
NMI 1-méthylimidazole NMP N-méthylpyrrolidone NMR nuclear magnetic resonance
NOE effet Overhauser nucléaire
NOESY spectroscopie nucléaire par effet Overhauser O
O ortho P
xviii Ph phényle
PL pantolactone
PNEA pantoylnaphtyléthylamine ppm partie par million
Pr propyle py pyridine Q QD quinidine QN quinine R
RMN résonnance magnétique nucléaire Rf facteur de rétention
rt temperature de la pièce (room temperature) S
s singulet (spectre)
SET transfert d’un seul électron
SN2 substitution bimoléculaire nucléophile STM microscope à effet tunnel
T
T température
t temps, triplet (spectre)
TBAF fluorure de tétrabutylammonium TBS (TBDMS) tert-butyldiméthylsilyle
TPS (TBDPS) tert-butyldiphénylsilyle
Tf trifluorométhylsulphonyle (triflyle) TFA acide trifluoroacétique
TFAA anhydride de trifluoroacétique TFAP 2,2,2-trifluoroacétophénone
THF tétrahydrofurane
TMEDA N,N,N’,N’-tétraméthyléthylènediamine TOF turnover frequencies
TON turnover number tp température de la pièce TPP tétraphénylporphyrine
TR temps de rétention (chromatographie) Ts tosyle
U
UHV ultra haut vide V VHC virus de l’hépatite C W w/w fraction massique X Y Z
xix
xx
L’imagination est, par suite, nécessaire au génie pour voir dans les choses non ce que la nature y a effectivement mis,
mais plutôt ce qu’elle s’efforçait d’y réaliser…
Arthur Schopenhauer, Le monde comme volonté et comme représentation Livre III. §36
I think nature's imagination is so much greater than man's, she's never going to let us relax.
xxi
Remerciements
En tout premier lieu : mon directeur John Boukouvalas : merci beaucoup de l’accueil au laboratoire. Tu as bien su pousser mes limites au-delà de mes propres attentes, tu m’as rendu plus fort, je t’en remercie et jamais je ne t’oublierai! À Peter McBreen : un privilège de t’avoir eu comme codirecteur. Ton équipe au laboratoire m’a permis de me développer et de m’accomplir tel que je le voulais. Je remercie mes deux directeurs de m’avoir donné la chance de faire de la recherche.
Merci aussi à tous les membres des groupes dont celui d’organique avec Richard pour l’admirable collaboration que tu m’as offerte dans les projets avec Peter et John (chapitre IIa et III). Vincent Albert, tu as été une source d’inspiration pour ton professionnalisme et j’ai adoré travaillé avec toi sur les projets des anhydrides (chapitre III). Charles, Marc-Alexandre, Ramesh, Lucas, Nicolas, Luiz, Karoline et Jaime, vous avez su me ramener à l’ordre lorsque mon enthousiasme faisait de l’écho dans le laboratoire. Thaís, tu as été une admirable collègue sur le projet avec les rubrolides (Chapitre V).
Merci à Vincent Demers-Carpentier, Guillaume, Yi, Yang, Caro et particulièrement Jean-Christian, avec qui j’ai vécu une excellente collaboration sur les projets avec les modificateurs chiraux (Chapitre IIb). Nous avons eu de bonnes aventures et partagé de bonnes conversations. La joie et l’enthousiasme allaient de pair aux travaux, surtout lorsque j’avais la chance de manifester mes compétences d’organicien devant une équipe de physiciens.
Remerciement chaleureux envers le laboratoire Paquin pour le HPLC et le GC chiral. Vous m’avez accueilli sans cesse pendant les nombreuses heures de mes analyses et j’ai bien aimé discuter avec vous.
Derniers remerciements à ma famille et mes collègues du département qui ont su me redonner l’énergie pour accomplir ce prestigieux projet d’aventure qu’est le doctorat.
1
2
La chiralité
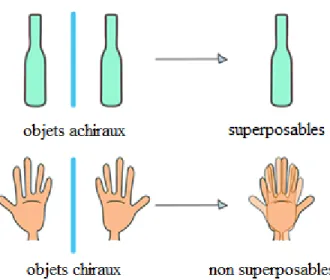
La chiralité est un cas particulier d’agencement spatial qui génère une distinction avec son image miroir. Autrement dit, c’est une propriété émergente de l’asymétrie, basée sur l’organisation géométrique des membres qui le constitue. Un exemple pour vulgariser ce phénomène est la main. Celle-ci est chirale et ne peut pas s’effacer en se superposant sur son image miroir (Figure 1).
Figure 1 : Présentation de la distinction des objets symétriques et des objets chiraux. Les objets achiraux sont superposables avec leur image miroir, mais pas les objets chiraux.
L’assemblage entre deux mains de même sens chiral forme une poignée, mais deux mains de chiralité opposée ne peuvent pas former cette même poignée. C’est le même comportement qui se produit dans la nature lorsqu’une molécule chirale rencontre une autre molécule chirale : la nature du lien dépendra de leur chiralité.
Une molécule chirale et son image miroir seront mises en relation par un terme nommé énantiomère. Cette paire d’énantiomères partage les mêmes propriétés physiques et chimiques, à l’exception lorsqu’elles vont interagir différemment avec des entités chirales, comme la lumière polarisée ou d’autres molécules chirales. Il faut considérer cette interaction comme un enjeu crucial, surtout lorsque le vivant est concerné. La chiralité est constamment
3
présente chez le vivant et son contrôle est donc nécessaire. À titre d’exemple, les organismes vivants se sont développés de façon homochirale avec les aminoacides dans les protéines et les sucres dans les structures à double hélice de l’ADN.1
Il est nécessaire de connaître le comportement avec le vivant de chacun des diastéréoisomères synthétisés. Ces études sont devenues une nécessité par souci de sécurité suite au danger inhérent à certaines paires d’énantiomères qui ont des effets différents dans le corps humain. Un des exemples est la molécule éthambutol : alors que l'énantiomère (S,S)-(+) est utilisable pour traiter la tuberculose, le (R,R)-(-) provoque la cécité (Figure 2).2
Figure 2 : Importance du contrôle de la chiralité dans les médicaments. L’exemple de l’éthambutol montre qu’un de ses énantiomères est un remède et l’autre est un composé qui provoque la cécité.
Il y a deux approches communes pour isoler des composés chiraux purs, aussi appelés énantiopurs. La première, c’est la séparation d’un mélange racémique. La seconde, ce sont des réactions qui convertissent des composés achiraux en composés chiraux.
Lorsqu’on peut transférer la chiralité sur une molécule achirale, celle-ci sera nommée prochirale, c’est-à-dire susceptible de voir apparaître une asymétrie dans sa nouvelle structure, créant ainsi une seule des images miroirs. Au cours du prochain chapitre, il sera exploré cet aspect de transfert de chiralité, particulièrement sur une surface faisant intervenir à la fois un composé prochiral et un système catalytique chiral. Ce système est composé d’un
4
ligand chiral fixé sur un catalyseur métallique. Dans notre cas particulier, les ligands sont des composés synthétiques inspirés d’un alcaloïde adsorbé sur une surface catalytique de Pt.
La catalyse
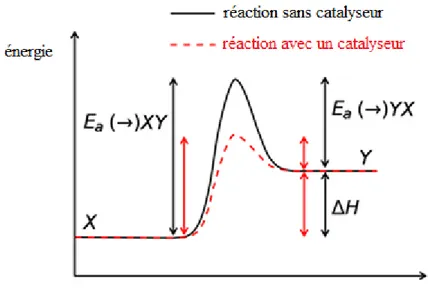
La catalyse est un procédé permettant d’accélérer une réaction à l’aide d’une substance appelé catalyseur. Il sert à abaisser l’énergie d’activation, changeant la vitesse de conversion des réactifs. Idéalement, c’est un composé qui n’est jamais détruit suite à la réaction, car il se régénère à la fin du cycle catalytique. La quantité nécessaire de catalyseur est donc plus petite que la quantité totale de substrat qui sera converti. Essentiellement, la conversion d’un produit en un autre peut être schématisée ainsi (Figure 3).
Figure 3 : Coordonnée de réaction pour la conversion du produit X en Y. Réaction non catalysée (noir). Réaction catalysée (rouge).
Pour convertir un produit chimique en un autre, il faut fournir l’énergie nécessaire au système réactionnel pour permettre aux composantes du produit X d’atteindre l’état de transition, représenté par le sommet de la courbe pour devenir le produit Y. Cette quantité d’énergie peut être abaissée grâce au catalyseur qui favorise la transition.
5
Les principaux procédés de catalyse sont des réactions de réduction et d’hydrogénation, d’oxydation, d’hydrolyse et de couplage C-C. Dans le particulier qui nous concerne, leur rôle du catalyseur d’hydrogénation est plus subtil. Il sert en plus de patron asymétrique, c’est-à-dire de pochette chirale. Cette pochette permet de contrôler la formation d’un seul stéréo-isomère. Le principe de ce contrôle est que le système catalytique chiral reconnaît une différence d’énergie de complexation entre les deux énantio-faces du composé prochiral. Cette différence peut être fortement contrôlée par des effets d’encombrement stériques et d’interactions attractives. Par ailleurs, le médium dans lequel il y a réaction définit le type de catalyse employé. Principalement, il y a deux types de catalyse : homogène et hétérogène.
La catalyse homogène
Lorsque le catalyseur se trouve dans la même phase que le substrat, la réaction sera catégorisée comme homogène. Parmi les avantages de la catalyse homogène, il y a une meilleure compréhension mécanistique due à l’accessibilité aux précatalyseurs et aux différents intermédiaires réactionnels, particulièrement, aux différents complexes que forme le catalyseur. Ces composés peuvent être isolés, analysés par les méthodes de spectroscopie et par la cristallographie.3
La catalyse homogène métallique
C’est un type de catalyse fine très employé dans le monde industriel, principalement dans le domaine agroalimentaire, pharmaceutique et des polymères.4 Parmi les catalyseurs les plus fréquemment utilisés on y retrouve la plupart du temps un catalyseur fait d’un complexe métallique avec des ligands phosphines. Ce type de catalyse d’une haute sélectivité s’utilise dans des conditions douces. Par rapport à la catalyse hétérogène, il n’y a pas les mêmes complications à la diffusion, donc au transfert de masse et aussi au transfert thermique, puisque la réaction se produit dans la même phase.
6
Voici un exemple (Schéma 1) de catalyse homogène métallique employé lors de la première synthèse de l’Antrocinnamomin D tiré du chapitre III. C’est un couplage C-C par catalyse au Fe.
Schéma 1 : Couplage croisé par catalyse homogène au fer lors de la synthèse de l’Antrocinnamomin D du chapitre III. La liaison C-C est formée entre le buténolide et le réactif de Grignard par la présence catalytique de l’acétylacétonate de Fe (III).
En 2010, la catalyse homogène métallique a été honorée par le prix Nobel en chimie pour ses prouesses en synthèse organique.5 Le prix a été partagé par les trois chercheurs de notoriété indéniable dans le domaine, soit Richard F. Heck, Akira Suzuki et Ei-ichi Negishi pour leurs travaux sur le couplage par le palladium. En voici un exemple du chapitre V, celui opéré dans la synthèse du rubrolide S (Schéma 2).
7
Schéma 2 : Couplage catalysé au palladium utilisé lors de la synthèse du rubrolide S. La formation du lien C-C entre le bromobuténolide et l’acide boronique est catalysée par le Pd(PPh3)2Cl2.
La catalyse homogène asymétrique
Au cours des dernières décennies, la catalyse homogène énantiosélective a fait des progrès phénoménaux, comme en témoigne le prix Nobel de 2001 remis à Sharpless, Noyori et Knowles pour leurs travaux associés aux hydrogénations et oxydations catalytiques chirales.6
Voici un exemple retrouvé dans la littérature (Schéma 3) en lien au chapitre IV. C’est la synthèse asymétrique d’une 4-hydroxy-1-tetralone grâce à un catalyseur de palladium hautement encombré par le ligand naturel (-)-spartéine. L’encombrement stérique permet de guider l’oxydation sur un seul des alcools du composé méso.7
8
Schéma 3 : Oxydation régiosélective par catalyse au Pd encombré par le ligand naturel (-)-spartéine lors de la synthèse énantiosélective d’une 4-hydroxy-1-tetralone à partir d’un diol méso.
La biocatalyse
La catalyse est fondamentale à la vie. Les réactions qui maintiennent la vie sont accomplies par des enzymes qui sont des catalyseurs naturels. Les enzymes sont principalement des macromolécules polypeptidiques composées de structures tridimensionnelles avec une pochette servant de site de liaison, réagissant avec une seule molécule réactive ou un groupe de réactifs. Cela qui signifie que les enzymes ont une capacité de reconnaissance moléculaire. La sélectivité est pratiquement de 100% et l’efficacité à réagir peut atteindre 10 000 molécules par enzyme par seconde, comparé à la catalyse hétérogène qui s’approche de 10 molécules par site par seconde. Les enzymes sont aussi très efficaces à des concentrations aussi faibles que 10-6 mol / L.8
L’utilisation des enzymes est la plus vieille catalyse industrielle alimentaire : ce processus enzymatique issu des micro-organismes est la fermentation. La nomenclature utilisée pour les enzymes combine deux termes : le type de réaction qu’elle accomplit et le suffixe –ase. On y retrouve six différents groupes : l’oxydoréductase, la transférase, l’hydrolase, la lyase, l’isomérase et la ligase pour respectivement l’oxydoréduction, le transfert de groupement fonctionnel, l’hydrolyse, l’addition ou la formation de liens doubles, l’isomérisation et la formation de liens. Leurs sites sont hautement spécifiques et peuvent différencier les isomères géométriques et optiques, réagissant avec un seul des énantiomères. Un exemple ici (Schéma 4) est une réaction d’oxydoréduction régiosélective d’une fonction
9
carbonyle par l’enzyme tétrahydroxynaphtalène réductase (T4HNR) issue d’un corps de champignon parasitaire appelé Magnaporth egrisea. Cette réaction se produit en présence du cofacteur NADP+ (Nicotinamide adénine dinucléotide phosphate) servant à la synthèse d’une autre 4-hydroxy-1-tetralone.9
Schéma 4 : Réduction énantiosélective par catalyse enzymatique d’un composé dione en 4-hydroxy-1-tetralone.
Cependant, les enzymes sont très sensibles au pH et à la température. De plus, la plupart ont des structures dont le rôle n’a pas encore été élucidé. Malgré tout, elles sont capables d’accomplir des réactions asymétriques d’une grande pureté.
Les catalyseurs homogènes sont en général fragiles, parfois très sensibles à l’air, nécessitant des conditions douces et ayant une faible stabilité thermique. Dans le cas des catalyseurs métalliques, ils sont constitués de métaux coûteux et parfois même de ligands encore plus coûteux, sans compter la difficulté de la récupération par séparation. En effet, récupérer un catalyseur homogène du milieu réactionnel est une complication puisqu’il se situe dans la même phase que le produit final. L’approche de fixer le catalyseur homogène sur une surface organique comme un polymère ou inorganique comme du verre ou de la céramique est possible. Cependant, cela diminue la flexibilité du catalyseur et de son accessibilité au substrat, ce qui affecte l’activité, sans compter les coûts additionnels de préparation et le contrôle de la densité de sites fonctionnels actifs sur la surface.8 Cette approche a pour but de faciliter les manipulations d’extraction du catalyseur de la phase. Ce type de catalyse existe et se nomme catalyse hétérogène.
10
La catalyse hétérogène
Par définition, la catalyse hétérogène fait intervenir plus d’une phase. Les grands avantages sont surtout d’un point de vue économique et technique, permettant une séparation facile du catalyseur et offrent la possibilité d’opérer en continu, très favorable pour l’industrie.10
Les catalyseurs hétérogènes typiques sont faits principalement de deux composantes. Il y a la partie active, qui est généralement un métal, soit sous forme native, d’oxyde, de sulfure ou de carbure. La deuxième composante est le support qui participe à la dispersion du métal sur la surface. Un exemple tiré du chapitre IV (Schéma 5) amplement employé dans l’académique est l’hydrogénation de fonctions insaturées, comme les oléfines et les alcynes par catalyse au Pd sur support de C.
Schéma 5 : Réduction par catalyse hétérogène d’un alcyne en alcane par hydrogénation sur Pd/C en présence d’hydrogène gazeux (50 psi).
La catalyse hétérogène métallique a dominé l’industrie pendant des décennies avant l’arrivée de la catalyse homogène métallique. Cette première possède une grande tolérance aux conditions de haute pression et de haute température. Les synthèses industrielles à grande échelle ne peuvent faire intervenir aussi facilement les mêmes procédés de manipulation et purification que ceux utilisés dans la recherche académique.
Parmi ces applications catalytiques, quelques procédés industriels sont devenus très utilisés. Un des plus connu, c’est de celui de la synthèse d’ammoniac à partir d’hydrogène et
11
d’azote gazeux. Le procédé se nomme Haber-Bosch et se fait à une haute température et pression sur un catalyseur de Fe avec un support d’alumine et d’oxyde de potassium.11 Un second procédé qui celui-ci est jumelé au procédé Haber-Bosch pour sa ressource d’hydrogène gazeux, c’est le reformage à la vapeur. C’est un procédé qui permet d’obtenir de l’hydrogène et du monoxyde de carbone, un mélange appelé syngas, à partir de méthane et d’eau sur un catalyseur de Ni.12 Il existe aussi le procédé inverse qui se nomme méthanation, permettant d’obtenir du méthane à partir du syngas sur une surface de Rh supporté.13 On peut aussi utiliser ce même syngas et le faire réagir sur un autre catalyseur hétérogène à base de Co, Fe ou de Ru, pour obtenir des hydrocarbures liquides et ce procédé se nomme Fischer-Tropsch.14 La catalyse hétérogène ne se limite pas qu’à ces exemples, il y a beaucoup d’autres, comme l’hydrogénation des huiles alimentaires, qui prennent aussi considérablement de l’espace sur le marché des catalyseurs hétérogènes.15
Ces procédés utilisent des conditions de pression et de température qui sont inappropriées pour des catalyseurs homogènes. Le fossé entre la délicatesse des catalyseurs homogène et les conditions sévères des catalyseurs hétérogènes font douter celles-ci d’être capable de livrer une marchandise fine comme la stéréo synthèse d’isomères, mais la recherche démontre qu’il existe des procédés hétérogènes qui sont capables de s’élever au contrôle énantiomérique actuellement dominé par la catalyse homogène asymétrique.
La catalyse hétérogène asymétrique
La catalyse hétérogène énantiosélective n’est pas au même niveau par rapport à la catalyse homogène en ce qui trait à la performance, à la connaissance mécanistique et à l’étendue du champ d’action des réactions possibles. Les seules réactions qui ont trouvé un succès applicable et un potentiel sont réduites à des hydrogénations asymétriques de carbonyle et d’oléfine.16
Le cheminement réactionnel typique pour ce type de réaction se fait dans cet ordre : il y a premièrement une adsorption du substrat prochiral en présence de l’auxiliaire chirale, ensuite il y a réaction sur la surface et finalement les produits quittent la surface par
12
désorption. La sélectivité de cette réaction dépendra non seulement du modificateur chiral, mais aussi de la géométrie des composés adsorbés sur la surface. Le modificateur chiral a la capacité de faire réagir préférentiellement avec une énantio-face, permettant d’obtenir le produit final avec la géométrie désirée. Cependant, l’inconvénient de cette catalyse est qu’elle dépend des différents sites actifs qui ne sont pas toujours réguliers, de la façon à laquelle le modificateur se fixe sur la surface et de la présence de différents sites non modifiés qui affecte le résultat final de la réaction en produisant des composés racémiques.
On répertorie trois cas de ce type de catalyse dans la littérature. Deux d’entre eux sont très étudiés. Le premier est la catalyse au Ni modifié avec l’acide tartrique, le second et troisième cas sont le Pd et le Pt modifiés par les alcaloïdes de la Cinchona, cette dernière aussi appelée la réaction d’Orito.
La catalyse au Ni
L’hydrogénation énantiosélective utilisant un catalyseur de Ni est la plus vieille réaction catalytique hétérogène énantiosélective connue.17 Cette réaction concerne principalement la réduction énantiosélective de cétones fonctionnalisées en β et les méthyles cétones encombrées sur une surface de Ni en présence d’acide tartrique. Cette réaction nécessite une haute température et pression d’hydrogène pour obtenir un bon excès d’énantiomère (Schéma 6).
Schéma 6 : Hydrogénation catalytique au Ni modifié par l’acide tartrique de cétones fonctionnalisées en bêta.
13
L’utilisation de l’acide tartrique R, R sur le Ni en présence du méthyle acétoacétate amène au produit de réduction R et on peut obtenir l’autre énantiomère du produit final en utilisant l’autre énantiomère de l’acide tartrique. Le plus grand excès d’énantiomère de 98.6% a été publié en 1997 pour l’hydrogénation du méthyle 3-cyclopropyl-3-oxopropanoate sur le Ni modifié et cette valeur reste la plus haute pour toute catalyse hétérogène asymétrique confondue.18
La catalyse au Pd
Cette catalyse hétérogène asymétrique est la plus récente et la littérature est moins abondante sur le sujet. Elle a démontré des activités modérées d’hydrogénation d’oléfine fonctionnalisée en présence d’alcaloïdes de la Cinchona sur une surface de Pd.19
La catalyse au Pt : la réaction d’Orito
La réaction d’Orito est une réaction catalytique hétérogène asymétrique d’hydrogénation de cétones activées. C’est un système catalytique reconnu d’une grande importance pratique et théorique pour la préparation énantiosélective de divers alcools (Schéma 7).
Schéma 7 : Hydrogénation énantiosélective de cétones fonctionnalisées en alpha à l’aide de Pt en présence d’un modificateur chiral, communément appelée la réaction d’Orito.
14
Elle possède la capacité de faire une réduction énantiosélective sur des types particuliers de substrats, incluant quelques cétones fonctionnalisées en α, tels des cétoesters, des cétoacides, des cétoamides, des dicétones, des céto acétals, des α-cétoéthers, des cétones avec une fonction trifluorométhane en alpha, des cétopantolactones et des pyrrolidine-2, 3, 5-triones.20 Cependant, la performance de cette réaction reste limitée par le manque de versatilité des substrats α-cétones activées.
Elle a été publiée pour la première fois en 1979 par Yoshio Orito et ses collègues.21 Originellement, c’est une réaction hétérogène qui fait intervenir un substrat prochiral alpha-céto ester. Ce substrat se fait réduire de façon asymétrique en présence d’hydrogène sur une surface de Pt avec un modificateur chiral qui peut être synthétique ou naturel comme les alcaloïdes de la Cinchona (Schéma 8). Le choix du solvant est crucial puisqu’il affectera la configuration modificateur-substrat sur la surface catalytique. Les solvants les plus rapportés par Alfons Baiker, un pionnier dans le domaine, sont l’acide acétique et le toluène. La pression du système est celle de l’hydrogène gazeux et doit être suffisante pour que l’hydrogène moléculaire se fixe sur la surface de Pt. La réaction peut s’opérer à des pressions d’hydrogène en deçà de 5 bar mais le prétraitement du catalyseur à 400 degrés est nécessaire pour obtenir un meilleur taux de réaction et excès d’énantiomère.22 Baiker et ses collègues pensent que ce prétraitement affecte la taille, la dispersion et la morphologie des particules et permet la suppression d’impureté sur la surface.23
Schéma 8 : Hydrogénation énantiosélective publiée par Orito. Conversion du benzoylformate d’éthyle en mandelate d’éthyle sur le Pt modifié par la cinchonidine.
15
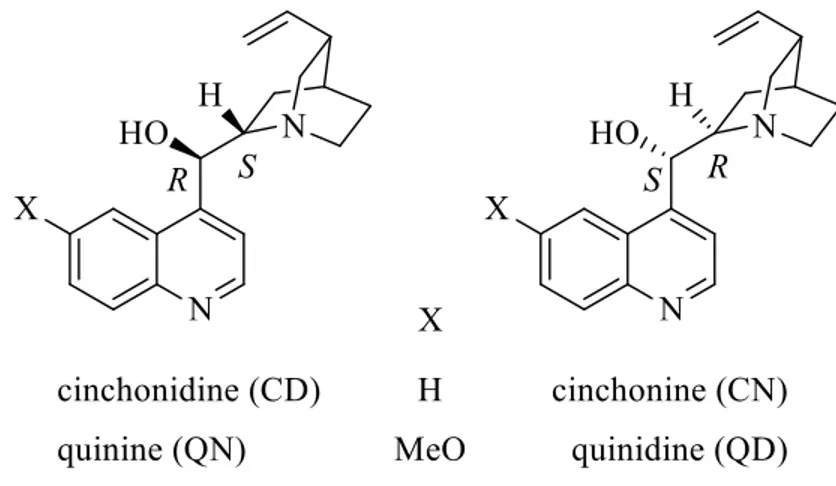
L’énantiomère S est obtenu à plus de 80% d’excès d’énantiomère avec la cinchonidine. L’autre énantiomère peut être obtenu avec un rendement optique semblable en utilisant la cinchonine. Ces modificateurs naturels, comme la cinchonidine et la cinchonine, sont des alcaloïdes issus d’une certaine variété d’écorces d’arbre de l’espèce Cinchona. Ils sont des produits naturels très accessibles à faible coût. Un de ces alcaloïdes chiraux, la quinine, elle a été isolée et purifiée pour la première fois par deux pharmaciens, Pierre-Joseph Pelletier et Joseph-Bienaimé Caventou en 1820.24 Louis Pasteur en 1853 l’a utilisé comme agent de résolution afin de séparer un mélange d’énantiomères de l’acide tartrique.25 Ces paires d’alcaloïdes pseudo-énantiomères comme la paire quinine/quinidine et la paire cinchonidine/cinchonine sont des métabolites secondaires herbicides naturels, aussi employés comme agents antipaludiques. Chaque alcaloïde a son pseudo-énantiomère, plus précisément son diastéréoisomère (Figure 4). La raison est qu’ils ont deux centres chiraux inversés et la même chiralité sur la fonction quinuclidine.
Figure 4 : Quatre pseudo-énantiomères extraits de la Cinchona. Les deux centres chiraux représentés distinguent les pairs CD/CN et QN/QD. Le groupement X (H, OMe) distingue les composés CN/CD des composés QN/QD. L’amine chiral bicyclique appelée quinuclidine est la même pour les quatre composés, empêchant ainsi les composés d’être de vrais pairs d’énantiomères.
Chaque membre d’une paire a la capacité d’agir comme modificateur dans la réaction d’Orito. Ce qui différencie les paires (QN, QD) et (CN, CD) est la présence de la fonction méthoxy sur l’anneau quinoléine.
16
Ces alcaloïdes sont très utilisés dans la littérature comme source d’énantiosélectivité dont la dihydroxylation asymétrique de Sharpless.26 L’équipe de Karl Barry Sharpless avait remarqué que ces alcaloïdes jouaient non seulement un rôle de transfert de la chiralité au cours de cette oxydation, mais aussi de catalyseur amenant une augmentation de la performance de cette réaction. Le terme qu’il a utilisé est l’accélération grâce au ligand et ce phénomène n’est pas étranger à l’influence que les modificateurs chiraux ont dans les réactions catalytiques hétérogènes énantiosélectives. Leur rôle est donc de rendre la réaction énantiosélective plus performante que la réaction racémique.
Cette dihydroxylation asymétrique fait intervenir un mélange commercial, soit un mélange partagé d’oxydant et d’auxiliaire chiral, appelé le mélange AD-mix, opérant en présence d’eau dans le tert-butanol et l’oxydation de la double liaison en diol se fait à l’aide d’un dérivé d’un de ces alcaloïdes (Schéma 9).
Schéma 9 : Synthèse du NED par dihydroxylation à partir du vinylnaphtalène avec le mélange Ad-mix bêta.
Ici, c’est le composé AD-mix β qui est employé, qui a servi pour synthétiser le modificateur chiral (R)-1-naphtyl-1,2-éthandediol ou NED. Nous avons utilisé cette méthode pour la synthèse de ce modificateur chiral qui a été étudié dans la seconde partie du chapitre II.27 Dans ce mélange AD-mix β, on y retrouve trois sels de potassium dont le carbonate, l’oxydant osmium tétroxyde et le réoxydant ferricyanure. Il y a aussi un ligand chiral, dérivé de la quinidine (Figure 5).
17
Figure 5 : Ligand chiral du mélange AD-mix bêta composé de deux molécules de dihydroquinidine et d’une molécule de phtalazine (représenté en bleu).
Ce ligand chiral est composé de deux molécules de dihydroquinidine liées à l’adduit phtalazine (PHAL). La quinine et la quinidine, bien qu’ils soient en relation de
diastéréoisomères dus aux autres centres chiraux identiques présents dans la fonction quinuclidine, agissent comme une paire d’énantiomères et chacun d’eux permettent d’obtenir au choix le bon énantiomère du produit chiral voulu, soit en employant l’AD-mix α, dérivé de la quinine et l’AD-mix β, dérivé de la quinidine.
Dans la littérature, on retrouve d’autres exemples de dérivés de la quinidine qui joue un rôle d’auxiliaire chiral employé pour guider de façon énantiosélective des réactions, comme dans le réarrangement de Kornblum-DeLaMare (Schéma 10).28
18
Schéma 10 : Réarrangement de Kornblum-DeLaMare asymétrique sur un composé diène avec l’utilisation d’oxygène singulet et de quinidine comme auxiliaire chirale.
La surface catalytique dans la réaction d’Orito est constituée de Pt, mais le modificateur joue aussi un rôle de catalyseur, en ce sens qu’il facilite la réaction chirale par rapport à la réaction achirale, soit celle sans modificateur. Comme mentionné plus tôt, un catalyseur a pour le rôle d’abaisser l’énergie d’activation permettant de convertir un réactif en produit. Le modificateur fournit un patron dans lequel le substrat prochiral est guidé et sera favorablement réduit d’un seul côté, donc amenant à un produit obtenu avec une énantiosélectivité. Idéalement, cette réaction devrait être unique, mais on y observe au mieux, une réaction majoritaire. Il se produit plus qu’une réaction, il y a donc des réactions secondaires, expliquant le résultat présenté en excès d’énantiomère. Les réactions secondaires minoritaires, responsables de fournir l’autre énantiomère, seront explorées dans la seconde partie du chapitre II.
En 1996, le groupe de Baiker a publié que le composé (R)-1-(1-naphtyl) éthylamine (NEA) s’est démontré très actif dans la réduction énantiosélective du pyruvate d’éthyle sur la surface de Pt, atteignant des excès d’énantiomère de 82%. En comparant avec la biomolécule cinchonidine (98% ee), il est évident que la complexité du composé naturel participe de façon optimale à obtenir un excès d’énantiomère comparable à celle des enzymes.29
19
Au cours de l’étude du NEA, ce groupe de recherche a observé que le modificateur chiral se modifiait in situ, amenant à un modificateur plus complexe, soit le (2R, 1’R)-(1’-(1-naphtyl) éthyl)-2-amino propionate d’éthyle, c’est-à-dire une amine secondaire, qui était responsable de l’excès d’énantiomère et que l’amine primaire NEA n’était qu’un précurseur.30 Cette hypothèse a ouvert la porte aux développements de nouveaux modificateurs, dont le pantoylnaphtyléthylamine (PNEA) avec sa structure initialement publiée (Figure 6). Le PNEA a sur son amine une structure dérivée d’un des substrats actifs, le cétopantolactone (KPL).31
Figure 6 : Quatre modificateurs chiraux synthétiques. Le NEA est le seul modificateur commercial. Le (2R,1'R)-[1'-(1-naphtyl)éthyl]-2-amino propionoate d'éthyle et le PNEA avec sa structure originellement publiée sont obtenus à partir du NEA.
Dans le cas d’une réaction comme celle d’Orito, le mécanisme reste irrésolu, pour la simple raison, comme l’indique la définition, que la réaction se produit sur plus d’une phase, soit à l’interaction des deux phases. Il est quand même possible d’isoler des intermédiaires
20
de réactions qui peuvent jouer un rôle dans la réaction. Ce point sera exploré dans la première partie du chapitre II.
Il faut opérer par la spectroscopie pour obtenir de l’information sur la structure des composés participant à la réaction sur l’interface des deux phases, soit, dans notre cas, celle de la surface métallique sur laquelle la réaction se produit. De plus, les sites actifs du catalyseur hétérogène ne sont pas toujours bien définis, bien que dans le cas d’Orito, la littérature mentionne que c’est la coupure (111) du cristal de Pt qui offre les meilleurs résultats (Figure 7).
Figure 7 : Trois coupures de cristaux selon l’indice de Miller. Les coordonnées en parenthèse représentent successivement l’interaction du plan avec l’axe des abscisses, l’axe des ordonnées et l’axe des cotes.
Le type de coupure du cristal résulte en une surface spécifique et influe sur l’énergie d’adsorption et l’activité catalytique. La surface (111) est celle qui offre les meilleurs sites actifs permettant d’obtenir le meilleur excès d’énantiomère.32 Il existe aujourd’hui certains appareils qui peuvent fournir de l’information à résolution atomique ou moléculaire sur les surfaces métalliques comme le Pt(111). Un de ces appareils, celui-ci appelé microscope à effet tunnel (STM), est utilisé par le laboratoire des surfaces de l’Université Laval.
21
STM
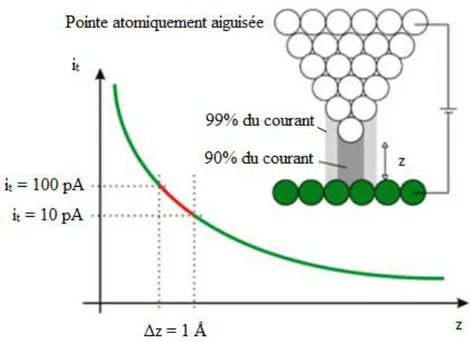
La microscopie par effet tunnel ou scanning tunneling microscopy (STM) est une technologie relativement récente. Son invention a gagné le prix Nobel en physique en 1986. La technique applique une propriété quantique permettant aux électrons de traverser une barrière de potentiel en exploitant un phénomène appelé effet tunnel. Cette microscopie offre une résolution à une échelle fractionnaire du nanomètre, soit 0,1 nm ou encore 1’Ångström.
Les électrons de valence d’un métal occupent un continuum d’états, nommé la bande de valence, jusqu’à une énergie nommée le niveau de Fermi (Ef). Ainsi, dans la bande de valence, les états en dessous du Ef sont occupés et les états au-dessus du Ef sont inoccupés. Dans l’expérience STM décrite dans cette thèse, une pointe atomiquement aiguisée de W est approchée très proche (de l’ordre d’angströms) de la surface du cristal du Pt(111). Un transfert d’électrons entre les deux métaux se produit naturellement, par effet tunnel, jusqu’à une égalisation de leurs niveaux de Fermi respectifs. Ensuite, si on applique une tension positive (ex : 1 eV) sur l’échantillon, on désaligne les Ef par 1 eV. Dans ces conditions, des électrons de l’énergie entre Ef et (Ef-1eV) du W pourraient faire un passage à travers le vide vers des niveaux inoccupés du Pt. Ainsi, on mesure un courant tunnel (Figure 8).33
22
Figure 8 : Jonction tunnel entre une pointe de W et la surface du platine. L’axe des abscisses représente la distance pointe-surface et l’axe des ordonnées le courant.
Ce courant décroit exponentiellement en fonction de la distance entre la pointe et la surface. Typiquement, un déplacement de la pointe de 1 Å plus loin de la surface entrainera une diminution du courant tunnel par un facteur de ~10. Étant donne cette sensibilité extrême à la distance pointe-surface, approximativement 99% du courant tunnel implique l’interaction de l’atome ultime de la pointe avec la surface. Ainsi, la pointe sonde la surface avec une résolution spatiale (x,y) à l’échelle atomique. Donc, par le déplacement fin en x,y de la pointe, en utilisant un tuyau piézoélectrique, il est possible, par exemple, de mesurer le courant tunnel sur une aire de 50 Å x 50 Å. Dans cette façon, la technique donne une image de la région sondée, où des variations en contraste révèlent des variations dans soit la structure soit la composition de la surface. Ainsi, la technique pourrait déceler la présence et la position et parfois l’orientation d’une petite molécule unique dans la région sondée.
Dans le cas qui nous concerne, le STM permet d’obtenir directement de l’information à une résolution moléculaire. Il est possible d’obtenir des images de type topographique d’un substrat adsorbé sur une surface de Pt(111). Les expériences sont directement inspirées des
23
conditions de réaction d’Orito, particulièrement de trois constituants de cette réaction catalytique hétérogène asymétrique : la surface, le modificateur chiral et le substrat.
Le STM (Figure 9) est sans doute la technique la mieux adaptée pour étudier des complexes uniques formés par une paire molécule chirale/molécule prochirale sur une surface. La technique est capable de discerner directement l’agencement du complexe. Ainsi, la technique permet, en principe, la détermination de la structure intramoléculaire du complexe diastéreomérique. Presque toute autre technique donne un signal venant de l’ensemble des molécules sur la surface.
Figure 9 : (a) Appareillage du microscope à effet tunnel (model : Specs Aarhus 150 STM). (b) Le scanneur y compris le tuyau piézoélectrique (5 et 9), la pointe (4) et l’échantillon (1).
Par contre, L’STM a également des limites sévères :
(i) D’abord, quoique la technique livre une résolution spatiale à l’échelle atomique, l’interprétation des images est loin d’être trivial. Cette difficulté découle du fait que l’image prise dans une mesure STM est, en effet, une carte de la densité locale d’états en x,y: c’est-à-dire l’image reflète la structure électronique pixel par pixel. Ainsi, il n’est pas évident de corréler l’image d’une molécule sur la surface à la structure de cette molécule. Cependant, à l’aide des calculs de la théorie de la fonctionnelle de la densité (DFT) et des mesures par spectroscopie infrarouge, il est possible de déterminer la structure du (R)-NEA sur le Pt(111)