Développement d’oligonucléotides antisens pour le
traitement de la dystrophie myotonique de Steinert
Mémoire
Dominic Jauvin
Maîtrise en biologie cellulaire et moléculaire
Maître ès sciences (M. Sc.)
Québec, Canada
III
Résumé
Le développement d’une thérapie génique pour la dystrophie myotonique de type 1 (DM1) implique l’utilisation d’un système de livraison musculaire efficace. L’évaluation d’oligonucléotides antisens (ASO) en conformation gapmer nous a permis d’identifier deux ASO, un de chimie 2’-O-méthoxyéthyle et l’autre avec des acides nucléiques bicycliques avec éthyle contraint, dont l’efficacité dans les modèles cellulaires et de souris de la DM1 était suffisante pour réduire significativement l’ARNm étendu de la DMPK. Il fut possible d’observer une réduction des foci nucléaires menant à une redistribution d’un régulateur d’épissage séquestré au noyau, ainsi corrigeant des erreurs d’épissage caractéristiques de la DM1. Plus particulièrement chez la souris DMSXL, l’injection systémique bihebdomadaire a mené à une maturation des fibres musculaires ainsi qu’au rétablissement de la force musculaire des sujets. Ce projet est la preuve de principe in vitro et in vivo qu’une thérapie génique par les ASO est concevable pour le traitement de la DM1.
V
Abstract
A gene therapy for myotonic dystrophy type 1 (DM1) implies an effective muscular delivery method. The evaluation of antisenses oligonucleotides (ASO) enabled us to identify two gapmer ASOs, one with a 2’-O-methoxyethyl chemisty and the other with a constrained ethyl bicyclic nucleic acid, whose efficacy in DM1 cell and mouse models was sufficient to significantly reduced expanded hDMPK mRNA levels. Furthermore, reduction in DMPK induced nuclear foci resulted in redistribution of a sequestered alternative splicing regulator, leading to correction of mis-splicing events characteristic of DM1. In DMSXL mouse, biweekly systemic injection of ASOs induced muscle fiber maturation and a gain in forelimb strength. This project is the in vitro and in vivo proof of the principle that an ASO gene therapy is conceivable for treatment of DM1.
VII
Table des matières
RÉSUMÉ... III ABSTRACT... V TABLE DES MATIÈRES ... VII LISTE DES TABLEAUX ... IX LISTE DES FIGURES ... XI LISTE DES ABRÉVIATIONS ... XIII REMERCIEMENTS ... XV AVANT-PROPOS ... XVII
INTRODUCTION ... 1
1. LA DYSTROPHIE MYOTONIQUE DE STEINERT ... 1
1.1HISTORIQUE DE LA PATHOLOGIE ... 1 1.2.SIGNES ET SYMPTÔMES ... 2 1.2.1. Forme tardive ... 2 1.2.2. Forme adulte ... 3 1.2.3. Forme infantile ... 3 1.2.4. Forme congénitale ... 3 1.3.L’ANOMALIE GÉNÉTIQUE ... 4 1.4.DIAGNOSTIC ... 5 1.5.MÉCANISME MOLÉCULAIRE ... 6 1.5.1. Physiopathologie de la maladie ... 6
1.5.1.1. Instabilité des répétitions à trinucléotides ... 6
1.5.1.1.1. Mécanismes d’instabilité ... 7
1.5.1.2. L’haploinsuffisance de la DMPK... 8
1.5.1.3. Haploinsuffisance des gènes adjacents à la DMPK ... 10
1.5.1.4. Toxicité de l’ARNm : modulation de facteurs d’épissage ... 11
1.5.1.4.1. Séquestration de MBNL ... 12
1.5.1.4.2. Élévation des niveaux de la protéine CUGBP1 ... 13
2. OUTILS MOLÉCULAIRES POUR UNE THÉRAPIE GÉNIQUE ... 15
2.1.STRATÉGIES THÉRAPEUTIQUES POUR LA DM1 ... 15
2.1.1. Élimination des ARN toxiques ... 15
2.1.2. Neutralisation de la toxicité des ARN ... 16
2.2.LES OLIGONUCLÉOTIDES ANTISENS ... 18
2.2.1. Les modifications chimiques ... 20
2.2.1.1. Première génération ... 21
2.2.1.2. Deuxième génération ... 22
2.2.1.3. Troisième génération ... 24
2.2.2. Pharmacocinétique des phosphorothioates ... 26
2.2.3. Toxicologie des phosphorothioates ... 28
3.1. HYPOTHÈSE... 31
VIII
DÉVELOPPEMENT ... 33
4.1.RÉSUMÉ ... 35
4.2.ABSTRACT ... 37
4.3.INTRODUCTION ... 39
4.4.RESULTS/DISCUSSION/CONCLUSION ... 43
4.5.METHODS ... 47 4.6.REFERENCES... 53 4.7.FIGURE LEGENDS ... 57 4.8.FIGURES ... 59 4.9.SUPPLEMENTARY INFORMATION ... 61 CONCLUSION ... 69 BIBLIOGRAPHIE ... 71
IX
Liste des tableaux
XI
Liste des figures
FIGURE 1-HANS STEINERT (1875-1911) ... 1
FIGURE 2-LOCUS DU GÈNE DE LA DM1 ... 4
FIGURE 3-LOCALISATION DES RÉPÉTITIONS EXPANSIBLES CAUSANT DES PATHOLOGIES ... 6
FIGURE 4-STRUCTURES FORMÉES PAR LES TNR DANS LA DM1 ... 8
FIGURE 5-GÈNE DE LA DMPK, SITES D'ÉPISSAGE ALTERNATIF ET DOMAINES STRUCTURAUX ... 10
FIGURE 6-COLLABORATION ENTRE MBNL1 ET CUGBP1 DANS LE CONTRÔLE DE L'ÉPISSAGE ALTERNATIF DURANT LE DÉVELOPPEMENT ... 14
FIGURE 7-PERTE DE FONCTION DE MBNL1 ET GAIN DE FONCTION DE CUGBP1 ... 14
FIGURE 8-MÉCANISME DE DÉGRADATION À LA RNASE H1 INDUIT PAR LES ASOS GAPMERS ... 20
FIGURE 9-MODIFICATIONS CHIMIQUES DES ASO ... 21
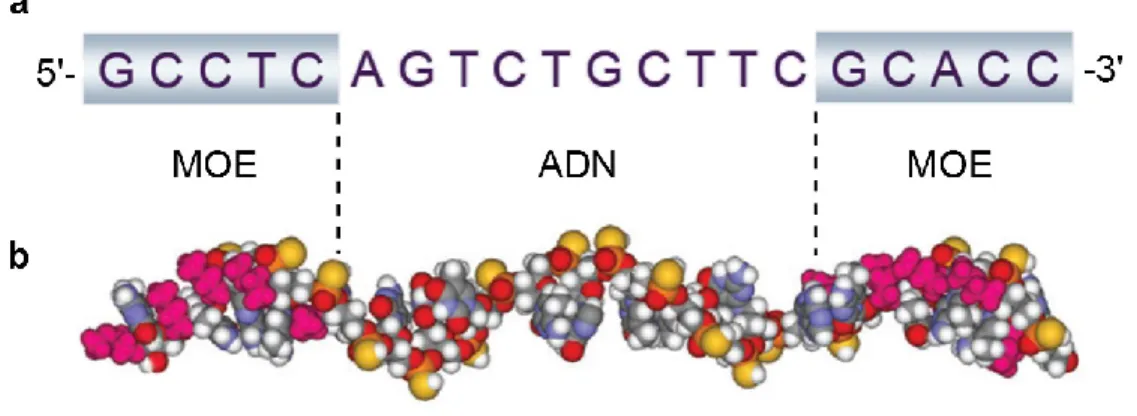
FIGURE 10-STRUCTURE D'UN GAPMER ... 23
FIGURE 11-MODIFICATION D'OLIGONUCLÉOTIDE (S)-CET ... 24
XIII
Liste des abréviations
AAV : « Adeno-associated virus » Virus adénoassociéADN : Acide désoxyribonucléique
ADNg : Acide désoxyribonucléique génomique
AGO2 : Argonaute 2
ALAT : Alanine aminotransférase
ARE : « AU-rich elements » Éléments riches en AU
ARNi : Acide ribonucléique interférent
ARNm : Acide ribonucléique messager
ASAT : Aspartate aminotransférase
ASO : « Antisense oligonucleotide » Oligonucléotique antisens
BNA : « Bicyclic nucleic acid » Acide nucléique bicyclique
CAG : Cytosine-adénine-guanine
CDM1 : « Congenital DM1 » Dystrophie myotonique de type 1 congénitale
CELF : « Elav-like family member » Famille des CUGBP ressemblant à Elav
cET : « 2’,4’-constrained-2’-O-ethyl » 2’-O-éthyle contraint en 2’,4’
CLCN1 : « Chloride channel 1, voltage-sensitive 1 » Canal chlore 1 voltage dépendant
CpG : Cytosine-phosphate-guanine
CPP : « Cell-penetrating peptide » Peptide d’incorporation cellulaire
CRIK : « Citron Rho-interacting kinase » Kinase citron interagissant avec Rho
CTG : Cytosine-thymine-guanine
CUG : Cytosine-uracile-guanine
CUGBP1 : « CUG binding protein 1 » Protéine 1 liant les CUG
DHPR : « Dihydropyridine receptor » Récepteur de la dihydropyridine
DM : Dystrophie myotonique
DM1 : Dystrophie myotonique de type 1
DM2 : Dystrophie myotonique de type 2
DMPK : « Dystrophia myotonica protein kinase » Protéine kinase de la dystrophie myotonique
DMWD : « Dystrophia myotonica WD-repeat containing protein » Protéine de la dystrophie
myotonique contenant des répétitions-WD
FDA : « Food and Drug Administration » Agence américaine des produits alimentaires et
médicamenteux
hDMPK : « Human Dystrophia myotonica protein kinase » Protéine kinase de la dystrophie myotonique humaine
hnRNP H : « Heterogeneous nuclear ribonucleoprotein H » Protéine ribonucléaire H hétérogène
INSR: « Insulin receptor » Récepteur à insuline
LNA : « Locked nucleic acid » Acide nucléique barré
mDMPK: « Mouse Dystrophia myotonica protein kinase » Protéine kinase de la dystrophie myotonique de souris
MBNL : « Muscleblind-like splicing regulator » Régulateur d’épissage comme muscle-aveugle
ME : 2’-O-méthyle
MEF2A : « Myocyte enhancer factor 2A » Facteur d’amplification de myocyte 2A
XIV
MRCK : « Myotonic dystrophy kinase-related CDC42-binding kinase » Kinase liant CDC42 en lien avec une kinase impliquée dans la dystrophie myotonique
MYPT1 : « Myosin phosphatase target subunit » Phosphatase à myosine ciblant la sous-unité
PCR : « Polymerase chain reaction » Réaction en chaîne par polymérase
PKC : « Protein kinase C » Protéine kinase C
PLM : Phospholemman
PLN : Phospholamban
PMO : « Phosphorodiamidate morpholino oligomer » Oligomer morpholino phosphorodiamidate
PNA : « Peptide nucleic acid » Acide nucléique peptide
PO: Phosphodiester
PS: Phosphorothioate
PPMO : « Peptide-linked phosphorodiamidate morpholino oligomer » Oligomer morpholino
phosphorodiamidate lié à un peptide
RISC : « RNA-induced silencing complex » Complexe de sous-expression induit à l’ARN
ROCK : « Rho-activated protein kinase » Protéine kinase associée à Rho
shRNA : « Short hairpin RNA » Petit ARN en épingle
siRNA : « Short interfering RNA » Petit ARN interférant
SIX5 : « Sine oculis homeobox homolog 5 » Homologue 5 de la boîte homéotique sine oculis
SMA : « Infantile spinal muscular atrophy » Atrophie musculaire spinale infantile
SP-PCR : « Small-pool polymerase chain reaction » Réaction en chaîne par polymérase de petits ensembles
TLR : « Toll-like receptor » Récepteurs semblables à Toll
Tm : « Melting temperature » Température de fusion
TNFα : « Tumor necrosis factor alpha » Facteur alpha de nécrose des tumeurs
TNNT2 : « Troponin T type 2 (cardiac) » Troponine T cardiaque
TNR : « Trinucleotide repeats » Répétition de trinucléotides
VSGGG : Motif d’acides aminés Valine-Sérine-Glycine-Glycine-Glycine
XV
Remerciements
La recherche exige non seulement une bonne compréhension théorique des phénomènes biologiques mais aussi une bonne maîtrise des techniques expérimentales, de coutume transmises par les professionnels de recherche et étudiants expérimentés. De ce fait, je souhaite remercier Jessina Chrétien et Laurie Martineau de leur contribution à ma formation au sein de notre équipe. Avoisinant notre laboratoire, plusieurs membres de l’équipe du Dr Jacques Tremblay, lui-même inclus, furent très accueillants et disposés à répondre à mes nombreuses questions. Plus précisément, merci à Joël Rousseau, Cathy Gerard, Dominique Ouellet et Pierre Chapdelaine, ce dernier soumis aux plus audacieuses de toutes mes questions.
Je me dois de souligner l’extrême patience et l’encouragement apporté par ma famille, pour qui j’ai été absent et préoccupé lors de cette entreprise un peu plus que je ne l’aurais aimé.
Ces travaux de maîtrise sont l’accomplissement d’une idée mignardée par le Dr Jack Puymirat depuis 10 ans déjà, que plusieurs caractérisaient de futile à l’époque. Grand visionnaire, il a mon respect et mon admiration pour son travail acharné qui lui a permis d’amener un oligonucléotide antisens en phase clinique I pour le traitement de la dystrophie myotonique de type 1. Je tiens à le remercier pour la confiance accordée à mon égard, pour la flexibilité dont il a fait preuve ainsi que pour son soutien dans l’accomplissement de cette maîtrise.
Finalement, je désire dire un gros merci à toute l’équipe chez Isis Parmaceuticals pour leur travail remarquable en vue de développer des nouveaux traitements pour de nombreuses pathologies ainsi pour que pour le soutien financier et matériel apporté à cette étude.
XVII
Avant-propos
Constrained Ethyl Modified Antisense Compound is a Promising Tool for Gene Silencing Following Systemic Antisense Treatment in Myotonic Dystrophy type 1
Cet article, dont je suis le premier auteur, sera soumis pour publication sous peu. J’ai conçu toutes les figures de l’article et effectué sa rédaction sous la direction du Dr Puymirat. Ma contribution dans la section in vitro fut les expériences d’hybridation in situ par fluorescence combinées aux immunocytofluoresces pour la détermination d’efficacité des ASO dans les cellules, pour la redistribution de MBNL1 et pour le compte des foci; les Northern blot; l’analyse des résultats des puces à ADN et l’analyse des résultats des évènements d’épissage alternatif dont la plateforme de Benoit Chabot a généré les résultats bruts. Laurie Martineau a effectué la totalité des expériences d’apoptose/nécrose in vitro ainsi que la vérification de la correction de l’épissage de LDB3. Étant donné la taille des expériences in vivo, Jessina Chrétien, Laurie Martineau et moi avons effectué conjointement les injections des souris, les tests de forces, la récolte de leurs tissus et organes, les extractions d’ARN, les transcriptions réverses et les RT-qPCR. J’ai effectué les analyses des résultats des puces d’ADN des souris traitées et Jessina a fait les immunohistofluoresces suivies des comptes des types de fibres musculaires. Guillaume Bassez et Lucile Revillod furent responsables des FISH sur les coupes de tissus, des comptes de foci sur celles-ci ainsi que de l’analyse de leurs résultats. Geneviève Gourdon et Aline Lachon ont généré la lignée de souris DMSXL et ont fourni plusieurs des sujets d’étude. Chez Isis Pharmaceuticals, Sanjay Pandey, Robert McLeod et Frank Bennett ont fait le triage initial des 3000 ASO et nous ont fourni les plus efficaces. Ils furent responsables de leur synthèse ainsi que de la sélection des séquences et des chimies utilisées.
1
Introduction
1. La dystrophie myotonique de Steinert
1.1 Historique de la pathologie
En 1876, la première maladie avec myotonie fut décrite par le Dr Julius Thomsen, par sa propre expérience et celle de sa famille avec la myotonia congenita1. Plus tard connu sous le nom de Thomsen’s
myotonia congenita, cette maladie génétique héréditaire cause de la myotonie et de l’hypertrophie musculaire, sans toutefois être une condition progressive. La myotonie est un retard à la relaxation musculaire après contraction, ce qui empêche le relâchement d’objets chez les personnes affectées2. Toutefois, cette condition n’est pas une dystrophie, car aucune faiblesse musculaire n’y est associée. À la suite de la publication de la maladie de Thomsen, une série de cas atypiques sont apparus, une myotonia congenita avec atrophie musculaire. Deux publications en 1909, une par Steinert et l’autre par Batten et Gibb, sont les premières où l’on associe clairement la dystrophie myotonique à une pathologie en elle-même. Étant donné la description détaillée des travaux de Steinert sur un total de 9 patients atteint de la maladie,
on la surnomma la maladie de Steinert dans l’Europe continentale pendant un temps, pour ensuite observer une prévalence à la dénomination : dystrophie myotonique ou dystrophia myotonica. Le médecin allemand (Figure 13) y faisait la description d’une condition beaucoup plus sévère et dégénérative dont les principales caractéristiques étaient l’atrophie et la faiblesse musculaire, une faiblesse faciale, la ptôse palpébrale et une atrophie sélective du sterno-cléido-mastoïdien à travers les muscles du cou. Il remarqua aussi une atrophie testiculaire chez 4 de ses patients, la première anormalité documentée à l’extérieur du système nerveux. Grâce à l’étude approfondie d’un cas
d’autopsie, Steinert observa une fibrose extensive, des changements dégénératifs dans plusieurs muscles squelettiques ainsi qu’une atrophie des fuseaux musculaires. Le prochain apport à la DM provient de Greenfield en 1911 qui, par l’étude d’une famille de 13 enfants, dont 5 atteints, découvre que 3 parmi eux sont infligés de cataractes. L’année suivante, Curschmann avance que les cataractes et la présence d’atrophie testiculaire seraient indicatives d’un problème endocrinien généralisé et que la DM devrait dorénavant être classifiée comme un désordre systémique généralisé. En 1918, de par l’historique médical des familles de 35 patients atteints de la DM et de cataractes, Fleischer a pu observer la récurrence des cas de cataractes et ainsi confirmer la nature héréditaire de la DM. Il est aussi le premier à suggérer le phénomène d’anticipation de la DM, soit la nature amplificatrice de la pathologie dans les générations subséquentes. Ce n’est qu’en 1948 que Bell et Thomasen effectuèrent des études systématiques auprès des familles d’individus affectés.
Figure 1 - Hans Steinert (1875-1911)
2
L’étude de 6 enfants présentant un historique de symptômes datant d’aussi loin que la naissance mena Vanier en 1960 à suggérer que certains des symptômes apparaîtraient in utero. De plus, il observe que leur condition pathologique ne progresse pas très rapidement: il vient d’identifier la forme congénitale de la maladie. La découverte du locus du gène causant la DM1 en 1971 fut le premier pas vers le décèlement de l’anomalie responsable de la condition1. Ce n’est qu’en 1992, par l’identification simultanée de plusieurs équipes, que l’on met la main sur la mutation causant la DM1, la présence d’une instabilité dans la répétition d’un trinucléotide CTG à l’extrémité 3’ non-codante du gène de la DMPK4,5. En bref, la fin du 20e siècle fut une période extrêmement précieuse quant à la caractérisation clinique de la maladie ainsi qu'à la découverte de sa base moléculaire.
1.2. Signes et symptômes
La DM est plus fréquente forme de dystrophies musculaires retrouvées chez l’adulte. La prévalence moyenne de la maladie à travers le monde, sans compter les régions à haute concentration, est de 1/10 0001. Toutefois, certaines régions isolées pâtissent d’un héritage génétique qui augmente l’incidence des maladies génétiques. C’est le cas de Charlevoix et du Saguenay-Lac-Saint-Jean, situés dans le nord-est du Québec, où un effet fondateur a résulté à une prévalence unique de 1/5506. La DM1 est une maladie autosomale dominante, ce qui implique qu’un parent atteint sur l’un de ses deux allèles aura 50% de chance de transmettre le gène de la DMPK étendu, et ce pour chaque enfant, quel que soit le sexe. Comme pour toutes maladies à répétition de trinucléotides, le nombre de répétitions corrèle négativement avec l’âge du début des symptômes et positivement avec la sévérité de la maladie7.
La DM1 est un désordre multisystémique affectant les muscles squelettiques, la fonction des muscles lisses, le système cardiorespiratoire, digestif, endocrinien, les yeux, le squelette, la peau, et le système nerveux central1. En fonction de l’âge d’apparition des premières manifestations, la maladie peut être classifiée d’un point de vue clinique en cinq formes: tardive, de l’adulte, de l’adulte jeune, infantile et congénitale.
1.2.1. Forme tardive
Les premiers symptômes apparaissent après 40 ans et la plupart du temps, les signes cliniques se limitent à des cataractes sous-capsulaires postérieures, une calvitie prématurée, de la myotonie et la possibilité de problèmes de conduction cardiaque8. Néanmoins, une étude sur 37 patients atteints avec un âge moyen de 55 ans révèle que 56% des individus étaient asymptomatiques9.
3
1.2.2. Forme adulte
Lorsqu’un individu est atteint de la forme adulte ou classique, on retrouve chez cette personne de 50 à 1000 répétitions CTG. Forme la plus fréquemment rencontrée, ces symptômes surgissent entre 20 à 40 ans et réduisent la longévité entre 48 à 60 ans. L’atteinte musculaire est caractérisée par une faiblesse distale, un facies myopathique avec ptôse palpébrale et la myotonie. Parmi les autres signes de la maladie, on retrouve des manifestations gastro-intestinales, cardiaques, pulmonaires, endocriniennes, de la fatigue, de l’apathie, un manque d’initiative ainsi que de la somnolence de jour. Les blocages de conduction et la tachyarythmie pourraient être responsables de 30% des cas de mortalité, le danger majeur de la DM110. Ces symptômes affectent sérieusement la qualité de vie des patients ainsi que celle de leur famille8. La forme adulte précoce se différencie seulement par une apparition des symptômes entre 10 à 20 ans.
1.2.3. Forme infantile
La forme infantile débute entre 1 mois et 10 ans et se caractérise par un nombre de répétitions en général plus important que dans les formes adultes. Il est habituellement possible de diagnostiquer cette forme par l’existence de faiblesses musculaires touchant le visage, de la myotonie et des problèmes cognitifs8. Parmi ceux-ci, on retrouve le déficit d’attention, l’hyperactivité, des désordres d’anxiété ainsi que des problèmes d’apprentissage associés au retard mental11. L’évolution naturelle et progressive de la maladie mène éventuellement vers les symptômes de la forme adulte.
1.2.4. Forme congénitale
Cette forme apparaît à la naissance et correspond à la présence de plus de 1500 répétitions CTG. Elle se caractérise par de l’hypotonie, de la faiblesse faciale, des troubles de déglutition, des problèmes respiratoires et est associée à un taux élevé de mortalité néonatale, 16% comparativement à 1,9% dans la population. Lors de la grossesse, elle peut être soupçonnée par une réduction des mouvements fœtaux et la présence de polyhydramnios, ce dernier causé par une difficulté de déglutition du fœtus. Quand les enfants survivent, ils sont affligés d’un retard moteur et mental1. Les études cliniques ont établi que dans la majorité des cas, la mutation responsable de la CDM1 provient de la mère. La raison de cette préférence n’est pas connue, mais on sait qu’elle n’est pas prérogative, car il y a quelques cas répertoriés de CDM1 transmise par le père12,13.
4
1.3. L’anomalie génétique
Vers les années 1950, des analyses génétiques quantitatives14,15 ainsi que des études familiales systématiques16,17 n’ont fait que confirmer ce que l’on savait déjà comme étant un fait, l’origine génétique de la DM1. Il a fallu près de 40 ans à partir du premier indice de corrélation génétique entre la DM1 et les groupes sanguins Luthérien jusqu’à l’identification de la mutation responsable de la maladie sur le chromosome 19q13.31 (Figure 2). C’est en 1992 que le travail se termina lorsque 3 équipes mirent au grand jour la séquence d’ADN complète du gène de la DMPK, comportant une séquence variable de trinucléotides CTG située dans la section 3’ non-codante du gène4,5,18. Dans les populations normales, le nombre de répétitions CTG dans le gène de la DMPK varie entre 5 à 37 dans les cellules sanguines, un polymorphisme à l’égard du nombre de répétitions. Par contre, lorsque ce nombre est de 38 à 49, on se trouve dans la zone des prémutations, ce qui implique que les générations futures auront possiblement plus de répétitions que la précédente. Malheureusement, ce phénomène induira des symptômes de la DM1 lorsque ce nombre dépasse 49.
Figure 2 - Locus du gène de la DM1
Le panneau de gauche situe le locus du gène de la DM1 sur une représentation schématique du chromosome 19. Le panneau de droite indique les gènes adjacents et les sondes utilisées pour localiser le locus du gène de la DM1. Illustration tirée de 19.
5
1.4. Diagnostic
Le diagnostic de la maladie repose essentiellement sur la notion d’hérédité et sur les signes cliniques. Dans les cas douteux, le diagnostic repose sur le test génétique qui a supplanté la caractérisation des biopsies musculaires (Tableau I).
Tableau I - Histologie musculaire dans la dystrophie myotonique de type 1
Caractéristiques principales:
Augmentation du nombre de noyaux centralisés Variation dans la taille des fibres musculaires Chaînes de noyaux
Fibres musculaires annelées Masses sarcoplasmiques Atrophie des fibres de type 1
Fuseaux musculaires : augmentation de la division des fibres Caractéristiques secondaires :
Petites fibres angulaires Fibres d’apparence « mitée » Hypertrophie des fibres de types 2 Augmentation de la fibrose
Le diagnostic moléculaire est effectué sur un prélèvement d’ADN du patient en provenance du sang, d’amniocytes ou de villosités choriales. La détection de la présence d’un allèle étendu du gène de la DMPK se fait par PCR pour les allèles sous 300 répétitions CTG. Dans les cas où leur taille est supérieure, l’amplification PCR sera impossible et aucun signal ne sera détecté. Le diagnostic est alors effectué par le Southern Blot sur de l’ADNg digéré, ce qui permet de déterminer toutes les tailles d’allèles DM1, soit de 5 à 4000 CTG.
6
1.5. Mécanisme moléculaire
1.5.1. Physiopathologie de la maladie
1.5.1.1. Instabilité des répétitions à trinucléotides
De nombreuses maladies neurodégénératives et neuromusculaires sont causées par l’expansion de répétitions de trinucléotides (TNR) située dans différentes régions des gènes (Figure 3). Cependant, on doit se demander comment un sujet normal transmet une incidence de DM1 ou un statut prémutatif par l’expansion des répétitions à ses enfants. Chez la levure comme chez l’homme, il fut démontré que les répétitions expansibles sont stabilisées par l’occurrence d’interruption à l’intérieur de la séquence répétée en prévenant la formation des brins glissés (Figure 4b). Pour l’instant, la théorie la plus probable est la perte de ces interruptions stabilisantes retrouvées à travers les répétitions20. L’instabilité résultante serait responsable de l’augmentation du nombre de répétitions lors de la transmission intergénérationnelle. Par conséquent, d’une génération à une autre, on constate un début de plus en plus précoce des prodromes et une sévérité accrue de la maladie, c’est le phénomène d’anticipation.
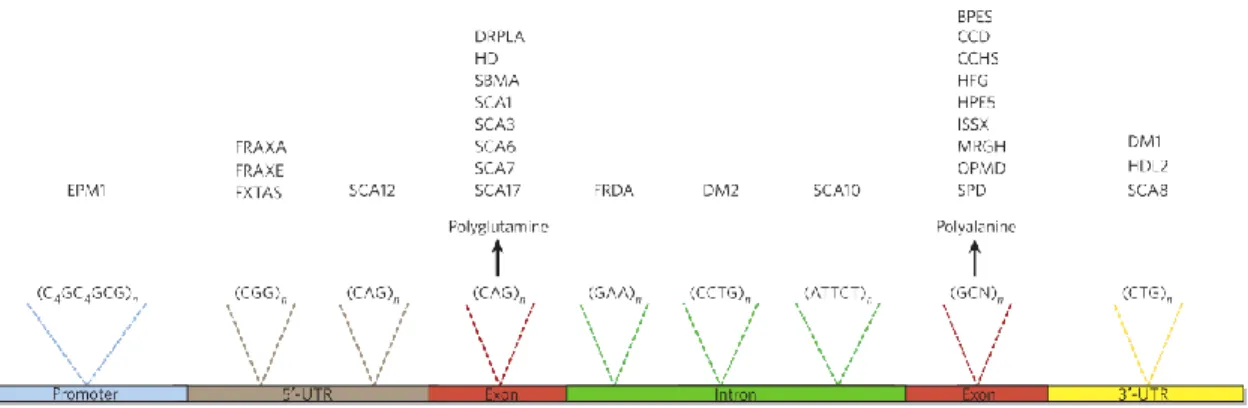
Figure 3 - Localisation des répétitions expansibles causant des pathologies
Séquence et localisation des répétitions expansibles causant des maladies humaines sur les différentes parties d’un gène générique. BPES, blépharophimosis épicanthus inverse et ptôsis; CCD, dysplasie cléidocrânienne; CCHS, syndrome d’hypoventilation congénitale; DM, dystrophie myotonique; DRPLA, l'atrophie dentatorubro-pallidoluysienne; EPM1, épilepsie myoclonique progressive 1; FRAXA, syndrome de l'X fragile; FRAXE, retard mental lié à l'X fragile associé au site FRAXE; FRDA, ataxie de Friedreich; FXTAS, syndrome du tremblement et d’ataxie de l’X fragile; HD, maladie d’Huntington; HDL2, maladie de Huntington de type 2; HFG, syndrome main-pied-génital; HPE5, holoprosencéphalie 5; ISSX, syndrome infantile des spasmes liés à l’X; MRGH, retard mental avec déficit en hormone de croissance; OPMD, dystrophie musculaire oculopharyngée; SBMA, atrophie musculaire spinale et bulbaire; SCA, ataxie spinocérébelleuse; SPD, synpolydactylie. Illustration tirée de 20.
7 Afin de mieux comprendre l’instabilité des répétitions dans la DM1, on a cherché comprendre la dynamique de transmission parentale des répétitions à travers les cellules germinales. Des analyses de SP-PCR sur des échantillons de sperme de patients atteints de la forme DM1 adulte ou prémutatif ont démontré la présence d’allèles étendus très hétérogènes quant à leur nombre de répétitions et une fréquence de mutation de près de 1 par gamète21. Du côté des gamètes femelles, les études démontrent une augmentation de la longueur des répétitions 10 fois plus élevée dans les oocytes que dans les spermatozoïdes d’individus atteints de la DM1 et ce, avec des allèles étendus de taille correspondante. C’est très probablement ce qui explique que la majorité des incidences de CDM1 ont un héritage maternel22. La différence d’instabilité dynamique des TNR de la DM1 entre les cellules germinales des mâles et des femelles n’est pas gouvernée uniquement par la longueur des répétitions, mais serait influencée par des facteurs spécifiques au sexe23.
Au cours du développement, les allèles étendus de la DM1 sont victimes d’une instabilité somatique qui n’est pas uniforme dans tous les tissus24-26. Ce mosaïcisme somatique dirigé vers l’expansion des répétitions progresse durant la vie d’un individu21,27,28. Les expansions retrouvées dans les muscles squelettiques21,25,26 et dans le cœur24 sont de 2 à 13 fois plus grandes que ceux des lymphocytes du sang périphérique79, et les plus petites sont situées dans le cortex frontal et le thalamus24. Par conséquent, un diagnostic effectué à partir d’un échantillon de sang pourrait mal refléter la concordance avec la nature et la sévérité des symptômes éprouvés par les patients29. Au sein d’un même tissu, on retrouve aussi une hétérogénéité des expansions des allèles étendus de la DM1, visible comme une bande diffuse ou une trace par analyse Southern Blot ou une bande pâle par SP-PCR21,27. De plus, les études longitudinales attestent de la relation entre la progression de l’hétérogénéité de la taille des allèles étendus vers expansions des CTG avec le temps28. L’instabilité somatique serait par conséquent un processus continu à travers la vie avec un biais vers l’expansion des répétitions23 et possiblement régulée différemment en fonction des tissus.
1.5.1.1.1. Mécanismes d’instabilité
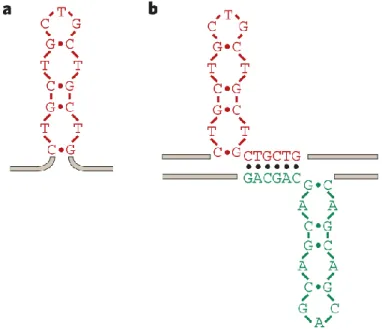
La première théorie tentant d’expliquer la présence de répétitions expansibles fut celle du glissement de l’ADN polymérase lors de la réplication de l’ADN30. Néanmoins, l’aire du séquençage à haut débit a remis cette explication en doute, car cette théorie n’expliquait pas pourquoi une petite proportion des répétitions dans le génome sont susceptibles à une expansion quand ce dernier est composé de 7,6% de répétitions en tandem31. Plusieurs modèles suggèrent des mécanismes expliquant la modification du nombre de répétitions dans la DM1 lors de la réplication de l’ADN, de la transcription, de la réparation, de la recombinaison, par des facteurs épigénétiques, et ce, souvent par la présence de structures intrinsèques aux TNR. Par exemple, les (CTG)n•(CAG)n successifs produisent des ânonnements de l’ADN polymérase menant à la formation d’épingle (Figure 4a) in vitro32,33 et in vivo34 , ce qui mène à une instabilité de la longueur des TNR (contraction ou
8
expansion) lors des processus cellulaires. Les épingles de TNR peuvent aussi être formées durant la transcription35 et lors de la réparation de l’ADN36 , ce qui entraîne la formation de structure de brins glissés37 (Figure 4b). Par conséquent, dès que l’on à la présence d’épingles sur les répétitions, il est possible qu’elles résultent en la modification de leur nombre lors de la réplication de l’ADN.
Figure 4 - Structures formées par les TNR dans la DM1
a. Malgré la complémentarité imparfaite des paires de bases de Watson-Crick, les répétitions expansibles (CTG)n•(CAG)n permettent
la formation de structures en forme d’épingle dans la DM1. b. Le glissement des brins peut entraîner la formation de structure de brins glissés dans les régions de répétitions. Illustration adaptée de 20.
1.5.1.2. L’haploinsuffisance de la DMPK
L’incapacité de trouver une explication unique quant à la cause des symptômes cliniques observés dans la DM1 guide l’opinion scientifique vers la possibilité de causes multiples. Les indices pointent vers les effets combinés d’une haploinsuffisance de la DMPK, de l’altération de l’expression des gènes adjacents et d’un effet pathogène des expansions CUG de l’ARNm de la DMPK. Néanmoins, on observe un consensus quant à la cause principale des symptômes de la maladie génétique en lien avec un gain de fonction toxique des transcrits des allèles étendus de la DMPK, causant une dérégulation de l’épissage alternatif. Les autres éléments responsables n’auraient que des rôles mineurs dans certains aspects de la maladie.
Suite à la découverte de la mutation de la DM1, on démontra que les niveaux d’ARNm et de protéine de la DMPK étaient diminués dans les biopsies musculaires de patients atteints de la DM1 adulte38. Ceci donna naissance à l’hypothèse d’haploinsuffisance, où une carence en DMPK induirait les effets attribués à la
9 pathologie. Toutefois, l’arrivée des modèles de souris est venue ébranler cette théorie. Les souris déficientes en DMPK ne démontraient pas les caractéristiques multisystémiques typiques de la DM1 et ne développaient qu’une myopathie tardive modérée39,40 et des défauts de conduction cardiaque41. Aucune myotonie de la part de ces souris, trait généralement associé à la maladie112. Quant à la surexpression de la DMPK par des souris
transgéniques, elle provoque une cardiomyopathie hypertrophique avec disrythmie, une myopathie myotonique et de l’hypotension, traits associés à l’atteinte musculaire de la DM139,42. La combinaison des effets de la modulation du niveau de la DMPK indique qu’elle peut contribuer à l’atteinte cardiaque et musculaire de la maladie, mais l’haploinsuffisance à elle-même n’explique pas la totalité des symptômes de la DM143.
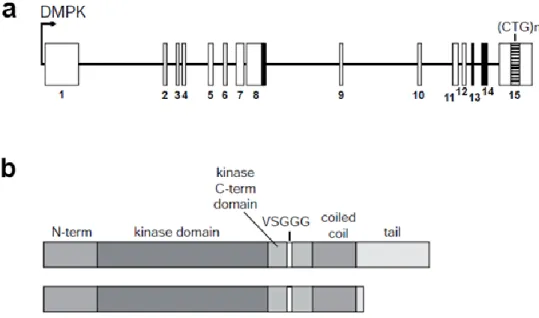
Le gène de la DMPK humaine et de la souris est très conservé44 et est constitué de 15 exons, dont les répétitions CTG se situent dans le dernier exon45 (Figure 5a). La protéine possède une taille théorique de 60 et 70 kDa pour les deux classes d’isoformes (Figure 5b) selon des travaux effectués sur de l’ADN complémentaire46. Toutefois, une seule isoforme fut clairement identifiée dans les tissus humains par un assortiment d’anticorps monoclonaux de souris et donne une taille réelle de 80 kDa47, augmentation causée par des modifications post-traductionnelles. L’épissage alternatif du transcrit primaire d’ARNm donne naissance à 6 isoformes majeures, désignées DMPK A à F46, et dont la spécificité des substrats dépend du type cellulaire et de leur localisation48. La présence d’une longue queue C-terminal permet l’ancrage de la DMPK dans la membrane externe mitochondriale ou dans la membrane du réticulum endoplasmique48,49 tandis que son absence chez les isoformes E et F induit une localisation cytosolique92. La DMPK fait partie de
la famille des protéines kinases de classe AGC incluant un total de 60 protéines classées en 21 sous-familles. C’est une protéine kinase activée par Rho (ROCK) comme la kinase citron interagissant avec Rho (CRIK) et la kinase liant CDC42 en lien avec une kinase impliquée dans la dystrophie myotonique (MRCKα/β/γ). Son rôle exact n’est toujours pas élucidé, mais on sait que ceux des protéines ROCK sont le contrôle de la morphologie et de la motilité cellulaire par le cytosquelette50. De par son domaine sérine/thréonine kinase (EC 2.7.11.1), on sait que la DMPK phosphoryle in vitro la sous-unité β de la dihydropyridine (DHPR)51, la phosphatase à myosine ciblant la sous-unité (MYPT1)52, la phospholemman (PLM) et la phospholamban (PLN)53. Outre le cerveau où l’apparition est plus tardive, l’ARNm de la DMPK est présent dans tous les types cellulaires d’origine myogénique à partir des phases de différentiation initiale chez la souris39.
10
Figure 5 - Gène de la DMPK, sites d'épissage alternatif et domaines structuraux
a. Schéma du gène de la DMPK et de ses 15 exons. Les boîtes blanches représentent les exons, la boîte lignée les répétitions CTG,
les lignes noires les introns et les boîtes noires les exons activement épissés. b. Les séquences communes aux isoformes de la DMPK sont le domaine N-terminal, le domaine sérine/thréonine kinase, le domaine C-terminal protéine kinase et le domaine d’hélices enroulées. Les domaines variables sont le motif VSGGG et le domaine C-terminal. Illustration tirée de 43.
1.5.1.3. Haploinsuffisance des gènes adjacents à la DMPK
La structure condensée de la chromatine dans la région du gène de la DMPK fut soupçonnée d’influencer l’expression des gènes adjacents, soit DMWD en amont et SIX5 en aval. Il fut remarqué que la présence des répétions CTG augmente l’efficacité de formation des nucléosomes et que leur présence pourrait réprimer la transcription dans cette région54. De plus, l’allèle étendu de la DMPK offre une résistance à la DNase I et une inaccessibilité aux nucléases ce qui confirmerait la présence d’une chromatine condensée55. Ceci dit, l’examen des niveaux d’expression de DMWD en lien avec la présence des répétitions donne des résultats contradictoires, car on observa sa baisse56,57 ainsi qu’aucun effet58,59. Le cas de SIX5 est beaucoup plus clair avec une diminution de 20-60% de son expression dans la DM156,58-60 corrélant avec l’augmentation du nombre de répétitions57. De plus, les répétitions CTG se retrouvent dans la région promotrice du gène SIX5, ce pourrait expliquer la diminution de sa transcription dans la DM1 si la chromatine de cette région est anormalement condensée. Chez la drosophile, ce gène code pour un facteur de transcription critique au développement musculaire, oculaire et testiculaire61. Un phénotype particulier chez les souris SIX5+/- est le développent des cataractes62,63, ce qui pourrait indiquer une légère contribution de SIX5 au phénotype de la DM1. Cependant, cette implication fut remise en question quelques années plus tard, lorsque le knockout de
11 MBNL1 chez la souris (MBNL1ΔE3/ΔE3), protéine à disponibilité réduite dans la DM1, développa des cataractes sans affecter les niveaux de SIX564.
1.5.1.4. Toxicité de l’ARNm : modulation de facteurs d’épissage
L’anomalie quant au nombre de répétitions a mené les chercheurs à vérifier si la présence elle-même des CUG sur l’ARNm a une implication dans la DM1. L’hybridation d’une sonde fluorescente (CTG)5 sur l’ARNm a démontré que celui-ci est bel et bien transcrit, mais qu’il est séquestré et s’accumule au sein du noyau sous la forme de foci65. Cette hypothèse fut testée chez la souris HSRLR, modèle comprenant 250 répétitions CTG dans la région 5’ non traduite du gène humain squelettique de l’α-actine. Le résultat fut très encourageant, car ces souris ont développé des inclusions ribonucléaires, la myotonie, une myopathie et des caractéristiques histologiques similaires à ceux de la DM166. Ceci démontra que l’unique présence de longues répétitions CUG est suffisante pour induire des symptômes propres à la DM1. Par la suite, les modèles de souris issus de la lignée DM300 (320 CTG) ont démontré le phénotype obtenu par l’incorporation d’une région du locus humain comprenant la DMPK ainsi que les gènes adjacents, pour un total de 45 kilobases. Cette lignée témoigne de l’instabilité des répétitions biaisées vers l’expansion, similaire à celle observée chez les patients DM1, et a donné naissance par instabilité intergénérationnelle à des lignées contenant des allèles étendus plus grands, comme la DMSXL avec plus de 1000 CTG. Par contre, pour une raison inconnue dans ce modèle de souris, on observe seulement les symptômes d’une DM1 légère à l’égard de la myotonie, d’une faiblesse musculaire et d’anomalies musculaires histologiques dans les souris âgées67. Il existe deux autres modèles de souris transgéniques inductibles à l’expression de 960 répétitions CUG dans la région 3’ non traduite de la DMPK qui récapitule les symptômes de la DM168,69.
Un élément qui nous a apporté une piste quant au mécanisme causal des symptômes par les répétitions est la découverte en 1998 d’une seconde forme de DM, la dystrophie myotonique de type 2 (DM2)70. Cette variante de la maladie est causée par la répétition d’un tétranucléotide CCTG dans le gène de la protéine 9 à doigt de zinc (ZNF9) et cause des symptômes similaires à la DM1, sauf pour l’absence d’une forme congénitale71. La ressemblance entre ces deux pathologies et la différence des gènes impliqués préconise un mécanisme pathologique commun, un gain de fonction toxique de l’ARNm. La présence d’une répétition de plus de 11 CUG favorise la formation d’une structure secondaire en forme d’épingle sur l’ARNm où les C-G sont séparés par des mésappariements de U-U72. Ces épingles d’ARNm double brin seraient ensuite capable de liées des protéines nucléaires, séquestrant ainsi les deux parties à l’intérieur du noyau. La soustraction de ces protéines aux mécanismes cellulaires sous un niveau critique induirait les symptômes de la DM1. Les protéines les mieux caractérisées quant à leur implication dans la DM1 sont la protéine 1 liant les CUG (CUGBP1) et les membres de la famille « muscleblind » (MBNL). La protéine ribonucléaire H hétérogène
12
(hnRNP H) est aussi capable de lier les répétitions lorsqu’en présence d’une branche d’épissage distale aux répétitions, et l’abolition de son expression par ARNi règle le problème de rétention nucléaire de l’ARNm de la DMPK73,74. On a aussi identifié des facteurs de transcription se liant aux transcrits de la DMPK étendue et par le fait même séquestrés, qui affecte les patrons d’expression génique75. L’étau se referme sur le fonctionnement d’un mécanisme pathologique très complexe et sa compréhension donnera certainement des indices sur celui des autres maladies à répétitions.
1.5.1.4.1. Séquestration de MBNL
La famille des protéines MBNL (MBNL1/2/3) fut initialement identifiée grâce à leur capacité de lier les répétitions CUG76 et est maintenant reconnue comme étant des régulateurs d’épissage alternatif. Les trois protéines furent détectées comme colocalisant avec les foci nucléaires d’ARNm étendu de la DMPK77 et cette localisation diminue radicalement leur abondance dans le nucléoplasme78, suggérant un mécanisme pathologique par séquestration protéique. Un modèle de souris sans le domaine de liaison à l’ARN (exon 3) de MBNL1 fut généré (MBNL1ΔE3/ΔE3) afin de déterminer l’implication de MBNL1 dans la DM1. Ces souris ont développé des traits caractéristiques de la DM1, comme les anomalies d’épissage, de la myotonie et des cataractes64. À l’inverse, la surexpression de MBNL1 par virus adénoassocié (AAV) dans les muscles squelettiques de souris HSALR corrige la myotonie ainsi que les erreurs d’épissage alternatif79,80. Durant le développement normal des muscles squelettiques, MBNL1 passe d’une localisation cytoplasmique à majoritairement nucléaire81. Il est donc suggéré que plusieurs des symptômes de la DM1 pourraient être causés par la séquestration de MBNL1 qui inverserait certains patrons d’épissage à ceux de leur correspondance embryonnaire82. Par exemple, l’épissage de la forme embryonnaire du canal chlore 1 voltage-dépendant (CLCN1) induit l’inclusion de l’exon 11 détecté dans la DM1. La présence de cet exon génère un codon-stop prématuré résultant à la sous-expression de la protéine CLCN1, qui fut démontrée comme étant la cause de la myotonie83,84. D’ailleurs, le traitement des souris HSALR par des morpholinos oligonucléotides antisens ciblant un site d’épissage sur l’ARNm de CLCN1 rétablit l’expression normale de la protéine et élimine les décharges myotoniques85. On observe aussi dans la DM1 un épissage anormal du récepteur à insuline (INSR), où l’absence de l’exon 11 diminue l’activité de signalisation tyrosine kinase86, produisant ainsi la résistance à l’insuline observée chez les patients87. La surexpression de MBNL1 dans des myoblastes DM1 fut démontrée comme suffisant pour corriger cette erreur d’épissage88. La troponine T cardiaque (TNNT2) subit aussi un défaut d’épissage, où l’augmentation de l’inclusion de l’exon 5 pourrait contribuer aux problèmes de conduction cardiaque89. Comme le témoignent ces résultats, on commence à établir une corrélation entre les erreurs d’épissage alternatif causé par la séquestration de MBNL1 et les symptômes de la DM1. Par contre, ce modèle n’explique pas encore perte de masse musculaire associée à la DM1 qui est d’ailleurs absente chez les souris MBNL1ΔE3/ΔE364.
13
1.5.1.4.2. Élévation des niveaux de la protéine CUGBP1
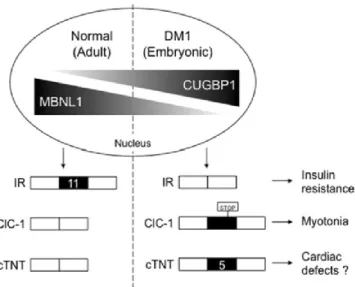
CUGBP1 fut la première protéine à capter l’attention des chercheurs dans la DM1 pour sa capacité à lier les CUG et les CCUG simple brin in vitro90. On a subséquemment découvert qu’elle ne se lie pas aux répétitions doubles brins en plus de ne pas colocaliser avec les foci d’ARNm de la DMPK91,92. Elle a toutefois été étudiée avec intérêt pour son niveau basal plus élevé dans les muscles89 et dans le cœur93 DM1. Les souris transgéniques surexprimant des répétitions dans la section 3’ non traduite du gène de la DMPK ont démontré que l’expression du transcrit muté est suffisant pour faire augmenter le niveau d’expression de CUGBP169. Lorsque surexprimé dans les muscles et le cœur de souris, CUGPB1 provoque des erreurs d’épissage et une mortalité néonatale94, supportant la théorie selon laquelle CUGBP1 contribuerait aux erreurs d’épissage observées dans la DM1. Un phénotype cardiaque de la DM1 est observé dans un autre modèle de souris lors d’une surexpression de CUGBP1 dans le cœur, en lien avec des changements fonctionnels, moléculaires et histopathologies décrits dans les modèles de souris DM195. CUGBP1 est l’une des six protéines de la famille des CUGBP ressemblant à Elav (CELF) dont le rôle familial est la régulation de plusieurs processus post-transcriptionnels, comme l’épissage alternatif, l’initiation de la transcription et la stabilisation de l’ARNm96-98. À partir de l’identification d’évènements d’épissage alternatif dans le cœur de souris par puce d’ADN, MBNL1 et CUGBP1 furent identifiés comme des régulateurs transcriptionnels dépendant du développement dans 24 cas d’épissage. Certains des évènements étaient modulés uniquement par MBNL1 (5) ou par CUGBP1 (13), et tous ceux régulés par les deux protéines (6) le sont de façon antagoniste99, comme en témoigne l’épissage de CLCN183, INSR87 et TNNT289. Durant le développement, le niveau de MBNL1 augmente tandis que celui de CUGBP1 diminue81, ce qui supporte l’hypothèse selon laquelle le niveau et la localisation de ceux-ci contrôle la transition de l’épissage fœtal vers adulte, situation qui se trouve anormalement inversée dans la DM182 (Figure 6). L’implication de CUGBP1 dans la DM1 fut associée à son rôle de substrat de la protéine kinase C (PKC), dont l’hyperphosphorylation et la stabilisation sont dépendantes de PKC activée. L’activation de PKC par un mécanisme inconnu produirait l’augmentation du niveau basal de CUGBP1, comme dans l’état embryonnaire100.
En résumé, le mécanisme pathologique privilégié pour la DM1 (Figure 7) est que la présence d’épingles de répétitions CUG séquestre MBNL1, ainsi diminuant sont niveau sous un seuil fonctionnel. Par conséquent, MBNL1 ne peut générer le patron d’expression adulte pour certains gènes. En même temps, un mécanisme inconnu non relié à la diminution de MBNL1 produirait l’augmentation de PKC activée. À son tour, PKC activée augmenterait le niveau basal de CUGBP1, responsable du patron embryonnaire de certains gènes causant ainsi la résistance à l’insuline et la myotonie. Étant donné la complexité de l’épissage alternatif combiné à la contribution de l’haploinsuffisance de la DMPK, il est probable que bien des années passeront avant une compréhension complète des causes des symptômes de la DM1.
14
Figure 6 - Collaboration entre MBNL1 et CUGBP1 dans le contrôle de l'épissage alternatif durant le développement
La transition entre les isoformes d’épissage alternatif embryonnaire à ceux adultes implique une expression différentielle de MBNL1 et de CUGBP1. Séquestré dans la DM1, MBNL1 ne peut favoriser l’isoforme adulte et l’on se retrouve avec des patrons d’épissages embryonnaires causés par l’augmentation des niveaux basaux de CUGBP1 phosphorylée. Illustration tirée de 82.
Figure 7 - Perte de fonction de MBNL1 et gain de fonction de CUGBP1
La séquestration de MBNL1 par une épingle formée de répétitions CUG diminue son niveau basal dans le noyau. Par un mécanisme inconnu, PKC est activée et phosphoryle CUGBP1 tout en augmentant son niveau de base. MBNL1 et CUGBP1 donnent ensuite naissance à une modification de l’épissage alternatif ainsi que des effets sur la traduction et la dégradation de l’ARN. Illustration tirée de 82.
15
2. Outils moléculaires pour une thérapie génique
2.1. Stratégies thérapeutiques pour la DM1
La compréhension des mécanismes moléculaires impliqués dans la régulation biologique donne espoir à la correction des défauts génétiques. À cette fin, la créativité de l’homme a donné naissance à plusieurs stratégies de traitements pour la DM1 basées sur la neutralisation/élimination des ARN toxiques. La toxicité provoquée par l’ARNm de l’allèle étendu de la DMPK a naturellement guidé la recherche vers une façon neutraliser et/ou de dégrader cet ARN, comme le témoigne les publications sur l’utilisation des oligonucléotides antisens (ASO), des petits ARN interférant (siRNA) et des ribozymes à tête de marteau autosectionnant. Par contre, deux stratégies principales sont présentement préférées: la première repose sur l’utilisation d’oligonucléotides antisens (ASO) qui se fixent sur les répétitions ainsi prévenant la liaison de protéines; la deuxième sur l’utilisation d’ASO qui se fixent sur l’ARN muté et induisent la dégradation des ARN toxiques.
2.1.1. Élimination des ARN toxiques
La destruction de l’ARN de la DMPK est visiblement l’approche la plus directe employée afin de tenter d’éliminer les effets causés par la toxicité de l’ARN. Un essai initial par Furling et al. (2003) a démontré que les ASO avaient le pouvoir de rétablir la fusion des myoblastes DM1 portant ~ 750 répétitions CTG ainsi que de normaliser leur incorporation du glucose par la correction du niveau d’expression de CUGBP1. L’ARN antisens était complémentaire aux (CUG)13 et exprimé dans les cellules par un vecteur rétroviral. Cette technique aurait préférentiellement réduit le niveau de DMPK muté de 80% par rapport à 50% pour l’allèle normal par un mécanisme non identifié101. La localisation exclusive de l’ARNm muté au niveau du noyau a ensuite inspiré Langlois et al. (2003) à utiliser un ribozyme à tête de marteau, aussi séquestré dans le noyau, afin d’essayer de cibler préférentiellement l’ARNm provenant de l’allèle muté. Cette technique a permis de réduire le nombre de foci nucléaires associé à la maladie et à corriger l’expression de l’isoforme B du récepteur d’insuline spécifique aux muscles, une erreur d’épissage caractéristique de la DM1102. Bien qu’encourageant, l’utilisation des ribozymes est freinée par la difficulté de prédiction de leur repliement in vivo103 et la complexité leur design à fait en sorte qu’ils ont été délaissés par d’autres technologies plus simples et efficaces, comme les ASO et l’interférence à l’ARN (iARN).
L’iARN implique l’utilisation de petits ARN doubles brins sous la forme de siRNA (petits ARN interférents) ou de shRNA (petits ARN en épingles). Une fois dans le cytoplasme, Dicer reconnaît ces petits ARN et les coupent en fragments de 21-25 nucléotides de long. Dans les deux cas, les siRNA résultants sont
16
chargés sur le complexe RISC où ce dernier utilise le brin antisens du siRNA afin de reconnaître les ARNm parfaitement complémentaires et de les couper grâce à l’activité catalytique d’argonaute 2 (AGO2). Dans le cas d’une complémentarité imparfaite incluant toutefois la région cible (« seed region »), l’évènement donnera naissance à une inhibition de la traduction104. Langlois et al. (2005) ont observé que l’expression de shRNA par des lentivirus dans des myoblastes DM1 humains portant ~ 750 répétitions CTG résultent en une diminution significative des niveaux d’ARNm de la DMPK provenant des deux allèles, y compris l’allèle muté séquestré dans le noyau, ce qui a des implications pour le mécanisme de l’interférence à l’ARN que l’on croyait agir seulement dans le cytoplasme105. Toutefois, l’approche des siRNA souffre d’une grande faiblesse, la méthode de livraison in vivo. L’utilisation de vecteurs viraux ouvre la porte à plusieurs dangers dont la mutagénèse d’insertion et à la dérégulation de l’expression génique104. L’administration systémique par voie parentérale est impossible à cause de la dégradation rapide des siRNA dans le sang par les nucléases. De plus, on ne peut pas compter sur des siRNA chimiquement modifiés, car le complexe RISC ne tolère que très peu de modifications chimiques sur le brin antisens. La prise en charge rapides des siRNA par le complexe RISC est donc essentielle à leur survie. La prise en charge des siRNA par le complexe RISC est très sensible aux modifications chimiques nécessaires à la survie des siRNA dans le sang et est restreinte à une administration locale ou jumeler à des substances favorisant leur livraison106.
En 2012, Lee et al. ont effectué la démonstration que l’utilisation d’ASO de chimie 2’-O-méthoxyéthyle (MOE) et acides nucléiques barrés (LNA) en conformation gapmer dirigés contre les répétitions CUG était capable de détruire environ 50% de l’ARN par un mécanisme dépendant de la RNase H lorsqu’administré par électroporation musculaire. Leur modèle était des souris inductibles à l’expression de l’exon 15 de la DMPK contenant 960 CTG107. La même année, Wheeler et al. ont publié dans le journal Nature que cette chimie MOE gapmer permettait, lorsqu’administré systémiquement chez la souris HSALR, une réduction de 80% de l’ARN du transgène hACTA1-CUGexp dans les muscles des pattes arrières de souris à 25 mg/kg 2 fois par semaine. Ils ont observé une diminution du nombre de foci nucléaires, une correction de la myopathie ainsi qu’une correction des erreurs d’épissage dépendant de MBNL1 : ATP2A1, TTN, LDB3 et CLCN1108.
2.1.2. Neutralisation de la toxicité des ARN
La neutralisation de la toxicité causée par les répétitions peut se faire par la liaison d’ASO chimiquement modifiés sur les répétitions CUG de l’ARNm de la DMPK, ce qui empêche l’interaction avec les facteurs de liaisons aux CUG. À cet effet, Mulder et al. (2009) ont utilisé un ASO 2’-O-méthyl-phosphorothioate qui a réduit de 90% le niveau d’expression du transcrit humain de la DMPK dans des myoblastes de souris DM500. Son efficacité in vivo était entre 30% à 80% dans les souris HSALR et dans les
17 DM500. De plus, leur ASO ciblait préférentiellement l’ARNm de l’allèle muté de la DMPK dans les myoblastes humains DM1, effet largement bénéfique pour une thérapie avec peu d’influence sur le niveau protéique de la DMPK. Dans les DM500, ils ont obtenu la réduction par un facteur 4 du nombre de foci et la correction de 5 évènements d’erreur d’épissage, soit ATP2A1, MBNL1, TTN, CLCN1 et TNNT3109. La même année, Wheeler et al. ont utilisé un ASO morpholino de 25 nucléotides qui a eu plusieurs effets bénéfiques lorsqu’injecté directement dans les muscles des souris HSALR : une diminution du nombre de foci, une redistribution de MBNL1 dans le noyau, une exportation des transcrits étendus de la DMPK dans le cytoplasme, une correction des erreurs d’épissage dépendantes de MBNL1, un retour à la normale de la conductance transmembranaire des ions chlores et une réduction de la myotonie110. Encore une fois dans le modèle de souris HSALR, Leger et al. (2013) ont testé l’utilisation des ASO morpholinos de 25 nucléotides liés à un peptide (PPMO) cationique dans le but de favoriser la biodistribution lors d’une administration par voie systémique intraveineuse. Leurs résultats ont montré une meilleure pénétration musculaire menant à la concentration suffisante pour corriger presque complètement les erreurs d’épissage, relarguer MBNL1 et éliminer de la myotonie111.
On a entrepris l’identification de molécules ou de substances modulaires multivalentes se liant aux répétitions CUG et qui pourrait empêcher la liaison de MBNL1 sur ceux-ci. Pushechnikov et al. (2009) ont mis au point un ligand perméable à la cellule dérivé du bisbenzimidazole Hoechst 3258 qui est capable de se lier aux répétitions CUG avec beaucoup d’affinité et de spécificité, empêchant la liaison de MBNL1112. D’autres équipes identifièrent la pentamidine et la néomycine B comme empêchant la liaison de MBNL1 aux répétitions. Néanmoins, seulement la pentamidine corrige des erreurs d’épissage dans des cellules HeLa exprimant 960 répétitions CUG ainsi que dans les souris HSALR. Des modifications devront être apportées à la molécule pour augmenter son affinité et sa spécificité afin de réduire la dose à administrer, car la dose efficace chez les souris était malheureusement mortelle113. Deux publications de Childs-Disney et al. en 2012 identifient l’approche de l’assemblage modulaire de molécules chimiques qui interagissent avec les répétitions CUG afin d’empêcher la liaison de MBNL1. Cette stratégie leur a permis de réduire la formation de foci in vivo et de corriger partiellement les erreurs d’épissage de ATP2A1 et CLCN1114. Wong et al. (2014) ont découvert un inhibiteur se liant dans les failles des hélices d’ARN qui permet d’empêcher la liaison de MBNL1 sur les CUG in vitro. Cette molécule, composée de deux triaminotriazines connectés par un bisamidinium, est perméable à la membrane cellulaire et au noyau. Elle diminue les foci et corrige est erreurs d’épissage (INSR et TNNT2) dans des cellules HeLa exprimant 960 répétitions CTG dans le gène de la DMPK. Cette molécule a aussi démontré une diminution de la toxicité induite par les répétitions dans un modèle DM1 chez la drosophile115. Ketley et al. (2014) ont développé un technique de criblage à moyen débit qui leur a permis d’identifié deux molécules chimiques, Ro 31-8220 et la chromomycine A3, qui éliminent les foci nucléaires, redistribuent et réduisent MBNL1 dans le noyau, normalisent CUGBP1 à son niveau basal et corrigent partiellement l’épissage
18
de INSR et ATP2A1. Le mécanisme fonctionnel de ces deux molécules n’est pas encore compris et nécessitera une étude plus approfondie116.
Les stratégies ciblant la réduction de l’ARNm de la DMPK sont rarement spécifiques au transcrit de l’allèle muté et pourraient avoir un impact sur les fonctions associées à la protéine. Afin de contrer ce problème, plusieurs voies n’impliquant pas le ciblage de l’ARNm furent élaborées. La première consiste à la modulation des niveaux protéiques de MBNL1 et CUGBP1, deux régulateurs de transcription débalancés dans la DM1. Dans un modèle DM1 de drosophile exprimant un ARNm non-codant contenant 480 CUG, Haro et al. (2006) ont surexprimé MBNL1, ce qui a eu pour effet de réprimer le phénotype dégénératif DM1 dans les tissus musculaires et les yeux. Étrangement, ils ont aussi observé une réduction du nombre de foci nucléaires, évènement non attendu pas cette stratégie en considérant notre compréhension du mécanisme pathologique de la maladie117. L’expression virale de MBNL1 dans les souris HSALR par Kanadia et al. (2006) a bien démontré la correction des évènements d’erreurs d’épissage associées à MBNL1 dans la DM1 (ATP2A1, CLCN1 et TNNT3) en plus de neutraliser la myotonie79. Dans le cas de CUGBP1, le rétablissement de niveau protéique basal par un blocage de l’activité de PKC dans un modèle de souris DM1 fut démontré comme étant une avenue potentielle de thérapie pour la DM1118. La modulation des facteurs d’épissage n’est pas une avenue sans danger, car l’épissage est un processus finement régulé dont la modulation grossière pourrait entraîner des effets indésirables. L’accumulation de l’ARNm étendu de la DMPK dans le noyau laisse présager qu’un rétablissement de l’exportation de ce transcrit dans le cytoplasme pourrait servir de traitement pour la DM1119. Cette avenue fut légèrement explorée, mais une compréhension plus approfondie du mécanisme d’exportation nucléaire de ce transcrit est nécessaire pour offrir une suite à cette stratégie120. La correction des évènements d’erreurs d’épissage par saut d’exons induit par des ASO est autre stratégie envisagée par certains chercheurs85. Cependant, étant donné le grand nombre d’erreurs à corriger, cette voie est probablement mieux adaptée d’autre pathologie comme la dystrophie musculaire de Duchenne, où l’excision d’un seul exon permet de rétablir le cadre de lecture normal de l’ARNm de la dystrophine menant à une protéine fonctionnelle121,122.
2.2. Les oligonucléotides antisens
La première réalisation du potentiel des acides nucléiques comme outil de régulation post-transcriptionnelle fut constatée lorsque Zamecnik hybrida un oligomère synthétique d’ADN de 13 bases à l’ARN 35S du virus du sarcome de Rous, bloquant ainsi l’initiation de la traduction et par conséquent la synthèse protéique123. La technologie résultante de cette découverte prit plusieurs années à se développer et demande des prérequis fondamentaux. La connaissance de la fonction et de la séquence de l’ARN que l’on veut affecter est nécessaire en vue de façonner la séquence de l’oligomère. Simple brin, l’ASO doit intégrer
19 l’organisme tout en restant intact pendant qu’il fait son chemin jusqu’à sa séquence d’ARN complémentaire ciblée. Afin d’augmenter leur capacité d’intégrer la cellule, ils peuvent être modifiés par l’ajout de substituants lipophiles, de ligand de récepteurs cellulaires ou de peptides d’incorporation cellulaire124. Le mécanisme par lequel les ASO dénichent leur ARN complémentaire à travers les centaines de millions d’ARN est toujours inconnu. Cette reconnaisse pourrait être tout à fait aléatoire par la simple diffusion de l’ASO à travers la cellule ou encore aidée par la prise en charge par une autre molécule. Habituellement d’une longueur de 15-20 nucléotides125, les ASO ont une spécificité remarquable qui leur permet de discriminer à la base près les ARN ciblés et de les lier avec un haut degré d’affinité126. Toutefois, en pratique, toutes les séquences ne peuvent être sélectionnées afin de créer des ASO efficaces. Tout d’abord, la séquence doit être unique afin d’éviter les interactions non désirées avec d’autres ARN. Ensuite, la complexité et la dynamique des structures secondaires et tertiaires que peut prendre l’ARN ainsi que la liaison de protéine sur celui-ci font obstacle à la reconnaissance. Des outils de prédictions structurales peuvent être utilisés pour réduire ces types d’interactions, mais ils ne sont pas parfaits127.
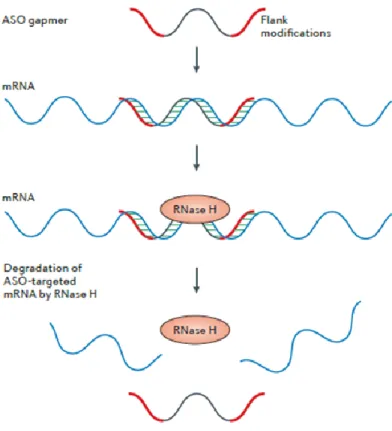
Le mécanisme de dégradation de l’ARNm par les oligonucléotides le mieux documenté est celui impliquant la RNase H1. Présente chez tous les mammifères, la RNase H1 est une endonucléase qui reconnaît spécifiquement les duplex d’ADN/ARN. Sa fonction habituelle au sein de l’organisme est la dégradation des amorces d’ARN synthétisé par la primase durant la réplication de l’ADN128. L’interaction entre la RNase H1 et les oligonucléotides serait rendue possible grâce à la formation de liaisons d’hydrogène entre l’enzyme et le squelette, et de nombreuses modifications chimiques diminuent ou abolissent ces liaisons129. Son activité de reconnaissance non spécifique nécessite un hétéroduplexe d’un moins 4 ribonucléotides afin de déclencher le clivage des liens phosphodiesters du brin d’ARN seulement130. Pour une application thérapeutique, ce mécanisme est très efficace, car l’insertion d’une seule molécule d’ASO donne naissance à plusieurs évènements de dégradation d’ARNm. À cet effet, le développement de la technologie des gapmers a su capitaliser sur l’efficacité de la dégradation de la RNAase H1 (Figure 8).
20
Figure 8 - Mécanisme de dégradation à la RNase H1 induit par les ASOs gapmers
La liaison d’un ASO de type gapmer permet le recrutement de la RNase H1 par sa région central et induit la dégradation de l’ARNm, libérant ainsi le gapmer pour un nouveau cycle de dégradation. Illustration adaptée de 106.
2.2.1. Les modifications chimiques
L’utilisation de molécules d’ADN ou d’ARN non modifiées à des fins thérapeutiques a fait face à certains problèmes intrinsèques qu’il fut nécessaire de régler afin d’offrir un avenir à la technologie des ASO. Tout d’abord, l’introduction d’ASO dans l’organisme induit très rapidement leur dégradation par les nucléases associées aux mécanismes de défense microbienne128, avant même de s’être rendues à leur cible. Ensuite, leur pharmacocinétique en fait une mauvaise stratégie systémique parce qu’ils sont rapidement filtrés du sang par les reins. Finalement, un système utilisant l’ADN pour cibler de l’ARN est victime d’une affinité inférieure à celle de l’ARN pour l’ARN. Pour ces raisons, les modifications chimiques des oligonucléotides furent explorées au niveau du squelette de phosphate, des hétérocycles, des sucres et ainsi que des stratégies de conjugaison afin de pallier à ces différents problèmes. Concrètement, les objectifs de ces modifications sont l’amélioration de la biostabilité, de l’affinité, de l’absorption cellulaire et de la distribution tissulaire des ASO131 tout en évitant au maximum la toxicité. Les efforts de synthèse ont donné naissance à trois générations de modifications chimiques (Figure 9).
21
Figure 9 - Modifications chimiques des ASO
La première génération d’ASO est caractérisée par la présence d’une modification chimique au niveau du squelette de phosphate où les liens phosphodiesters entre les acides nucléiques sont remplacés par des liens phosphorothioates (PS). La seconde génération provient de modifications 2’-alkyle sur le furanose et la troisième génération de modification provient généralement de modification de l’anneau de furanose. Illustration adaptée de 132.
2.2.1.1. Première génération
La première modification significative des ASO fut au niveau du squelette de phosphate, élément donnant la charge négative communément attribuée aux acides nucléiques. Les squelettes d’oligonucléotides contenant des liens phosphorothioates (PS) entre les nucléotides sont une des premières modifications chimiques efficaces effectuées. C’est la substitution d’un atome d’oxygène, ne participant pas à la liaison du phosphate entre deux nucléotides, par un atome de soufre qui différencie ce squelette des oligonucléotides normaux (Figure 9). Cette modification conserve la charge négative du squelette et confère aux ASO les