CRISPR-Cas9 pour l’édition de génomes viraux et
l’étude des gènes du phage virulent p2
Thèse
Marie-Laurence Lemay
Doctorat en microbiologie
Philosophiæ doctor (Ph. D.)
Résumé
Tous les écosystèmes contiennent des phages, ces virus qui infectent spécifiquement les bactéries. Les écosystèmes microbiens des produits laitiers ne font pas exception. Malgré les nombreuses recherches dans le domaine, les phages virulents spécifiques aux souches de
Lactococcus lactis utilisées pour la fermentation du lait menacent encore la qualité des
fromages et la constance des lots de production. Le phage p2 est un modèle pour l’étude des phages virulents de lactocoques, mais près de la moitié de ses gènes codent pour des protéines aux fonctions encore inconnues. L’étude des phages virulents constitue un défi de taille puisque la modification de leur génome est limitée par le court passage du génome viral dans la cellule bactérienne.
Le premier objectif de cette thèse était d’adapter un outil génétique basé sur la technologie CRISPR-Cas9 afin d’inactiver des gènes d’intérêt du phage p2. Cette technologie est dérivée d’un système antiviral naturel qui permet à certains procaryotes de se défendre contre l’invasion par de l’ADN étranger. La bactérie hôte du phage p2, L. lactis MG1363, est normalement dépourvue de ce système. Le deuxième objectif était d’étudier les protéomes phagiques et bactériens lors de l’infection virale par des analyses de spectrométrie de masse à haute résolution. Enfin, le troisième objectif était d’étudier les rôles des gènes inactivés sur la multiplication des phages et des bactéries infectées, incluant l’impact sur leurs protéomes. Entre autres, par une approche intégrative combinant des analyses génomiques, phénotypiques et protéomiques, j’ai comparé le phage mutant p2∆47, dont le gène orf47 avait été inactivé, au phage sauvage p2. Ces analyses m’ont permis de formuler une hypothèse quant à la fonction de la protéine virale ORF47.
Les phages sont ubiquitaires, abondants et peuvent se multiplier rapidement. Malgré leur importance et plus d’un siècle de recherches, plusieurs aspects de la biologie des phages demeurent mal compris. En concevant un outil pour la modification des génomes de phages virulents et en optimisant des protocoles d’analyses protéomiques, j’ai développé des méthodes efficaces pour la caractérisation des protéines phagiques et pour l’étude des interactions phage-bactérie.
Abstract
Phages are viruses that specifically infect and kill bacteria. They can be found in every ecosystem, including milk products. Despite decades of research, virulent phages infecting
Lactococcus lactis strains used for milk fermentation still threatens the production process
and cheese quality. Phage p2 is a model for the study of virulent lactococcal phages, but almost half of its genes encode proteins of unknown functions. The study of virulent phages is a challenge in itself because the modification of their genome is limited to the short infection cycle within a bacterial host.
The first objective of this thesis was to adapt a CRISPR-Cas9-based genetic tool to inactivate genes of interest in the genome of phage p2. The CRISPR-Cas9 technology is derived from a natural antiviral system that allows some prokaryotes to defend themselves against invasive nucleic acids. The bacterial host of phage p2, L. lactis MG1363, is naturally deprived of this system. The second objective was to study the viral and bacterial proteomes during phage infection, making use of high throughput mass spectrometry-based proteomics. Lastly, the third objective was to study the roles of inactivated genes on phage replication and bacterial growth, including the impact on their proteomes. Amongst other, with an integrative approach combining genomic, phenotypic and proteomic analysis, I compared the mutant phage p2∆47, lacking a functional orf47 gene, to the wild-type phage p2. These analyses allowed me to hypothesize about protein ORF47 function.
Phages are ubiquitous, abundant and can replicate quickly. Despite their importance and over a century of research, many aspects of phage biology are still poorly understood. By designing a tool for the modification of virulent phages and by optimizing protocols for proteomic analysis, I developed a robust pipeline to investigate uncharacterized phage proteins and to study phage-host interactions.
Table des matières
Résumé ... ii
Abstract ... iii
Table des matières ... iv
Liste des figures ... viii
Liste des tableaux ... x
Liste des abréviations ... xi
Remerciements ... xiv Avant-propos ... xvi Introduction ... 1 Lactococcus lactis ... 1 Caractéristiques principales ... 1 Importance de la souche MG1363 ... 2 Les phages ... 3 Le cycle de multiplication ... 3
Les protéines non structurales ... 5
Les phages dans l’industrie laitière ... 6
Le phage modèle p2 ... 8
Genetically modified and irradiated food: Controversial issues – facts versus perceptions ... 13
Résumé ... 14
Abstract ... 15
Glossary ... 16
Introduction ... 17
The microbial origin of CRISPR-Cas9 ... 19
Programmable DNA cleavage ... 20
Specificity ... 22
Repairing the cut ... 23
Plant transformation ... 26
Conclusion ... 29
References ... 30
The CRISPR-Cas app goes viral ... 35
Highlights ... 36
Abstract ... 38
Introduction ... 39
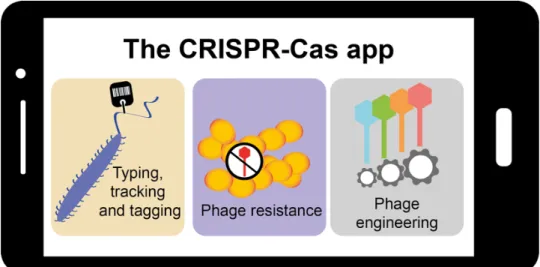
Strain typing, tracking, and natural genetic tagging ... 41
Phage resistance ... 41
Engineered phage therapy ... 44
Conclusion ... 49
Acknowledgements ... 49
References ... 50
Problématique, hypothèse et objectifs du projet ... 54
Chapitre 1 – Article 1 ... 55
Résumé ... 56
Abstract ... 57
Introduction ... 58
Results and discussion ... 61
Introduction of CRISPR-Cas9 into L. lactis MG1363 ... 61
Efficiency of pNZCas9 and pL2Cas9 protection against phage infection ... 61
Sequence analysis of escaping phages ... 63
Genome engineering of the virulent phage p2 ... 63
Targeting other phage p2 genes ... 66
Conclusion ... 70
Methods ... 71
Reagents and enzymes ... 71
DNA sequencing and analysis ... 72
Construction of pNZCas9 ... 72
Construction of pL2Cas9 ... 72
New spacer cloning in pNZCas9 and pL2Cas9 ... 73
Construction of homologous recombination templates ... 73
Analysis of recombinant phages ... 75
Supporting information ... 76 Acknowledgments ... 76 References ... 77 Chapitre 2 – Article 2 ... 80 Résumé ... 81 Abstract ... 82
In brief ... 83
Highlights ... 83
Introduction ... 86
Experimental procedures ... 88
Phage propagation ... 88
Time-course infection and protein extraction ... 88
GeLC-MS/MS ... 89
Targeted MS ... 90
Bacterial gene knockout using CRISPR-Cas9 ... 91
Analysis of L. lactis MG1363 recombinants ... 92
Data processing ... 93
Experimental design and statistical rationale ... 94
Results ... 95
Sensitivity of the method ... 95
Detection of a new phage protein ... 95
Detection of missing proteins ... 97
Visualization of host protein abundances during infection ... 98
Genome editing with CRISPR-Cas9 ... 101
Discussion ... 103
Uncovering phage p2 proteome ... 103
Investigating metabolic pathways triggered by phage p2 ... 103
Exploiting proteomics to design phage-resistant bacterial strains ... 105
Conclusion ... 108 Acknowledgments ... 109 Data availability ... 109 Supplemental data ... 109 References ... 110 Chapitre 3 – Article 3 ... 114 Résumé ... 115 Abstract ... 116 Introduction ... 117
Results & discussion ... 119
Sequence and structure homology ... 119
Impact of orf47 deletion on bacterial physiology ... 120
BIMp2∆47 genotype ... 122
Phage p2∆47 with an altered endolysin can infect BIMp2∆47 ... 122
Impact of ORF47 on bacterial proteomes ... 123
Conclusion ... 128
Methods ... 129
Structural analysis ... 129
Bacterial growth conditions ... 130
Phage propagation and titration ... 130
Complementation and bacterial gene knockout ... 131
DNA sequencing and analysis ... 132
Proteomic analyses ... 132
Acknowledgements ... 133
Supplemental data ... 133
References ... 134
Conclusion ... 136
Pourquoi étudier les phages? ... 136
Mutagenèse ... 137
Protéomique ... 139
Mécanismes de résistance ... 141
Accomplissements et perspectives ... 143
Références ... 146
Liste des figures
Figure 1. Schématisation du cycle de multiplication des phages virulents ... 5
Figure 2. Représentation du génome de p2 ... 9
Figure 3. Changement de conformation de la plaque basale de p2 en présence de calcium ... 10
Figure 4. Cas9 site-directed nuclease ... 21
Figure 5. DNA repair mechanisms ... 24
Figure 6. Base editing with dCas9 ... 25
Figure 7. Plant transformation ... 28
Figure 8. Graphical abstract for “The CRISPR-Cas app goes viral” ... 35
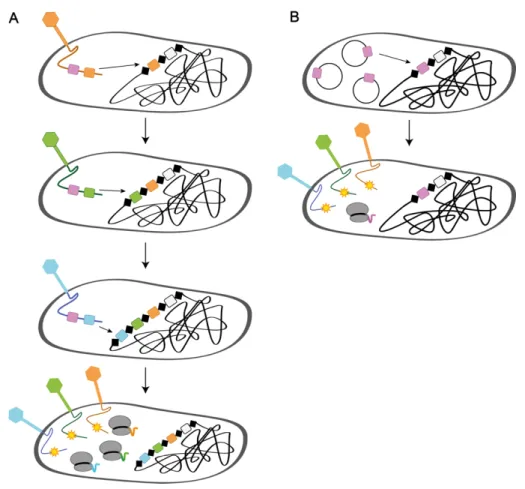
Figure 9. Generation of phage-insensitive mutants ... 43
Figure 10. A phage-transferable CRISPR-Cas9 system to kill or resensitize MDR bacteria ... 45
Figure 11. A cocktail of two phages to sensitize and lyse antibiotic-resistant bacteria ... 47
Figure 12. Graphical abstract for “Genome engineering of lactococcal phages using CRISPR-Cas9” ... 55
Figure 13. New spacer cloning in pL2Cas9 ... 62
Figure 14. Construction of three homologous recombination templates ... 65
Figure 15. Targeted genome editing of virulent phage p2 ... 67
Figure 16. PCR-based confirmation of deletions in the genome of phage p2 recombinants ... 69
Figure 17. Graphical abstract for “Investigating Lactococcus lactis MG1363 response to phage p2 infection at the proteome level”Abbreviations ... 84
Figure 18. Proteins detected in this study ... 96
Figure 19. Viral proteins detected during time-course infections ... 97
Figure 20. Expression profile of the phage p2 protein encoded by orf46 ... 98
Figure 21. Voronoi treemaps depicting the proteotypes of L. lactis cells during phage p2 infection ... 100
Figure 22. Growth curves of phage-sensitive L. lactis pKO0219 pL2Cas9 (control) and derivative L. lactis MG∆0219 in presence or absence of phage p2 ... 102
Figure 23. ORF47 overall fold in ribbon representation ... 120
Figure 24. Frequency of natural BIMs generated following the infection by either phage p2 or phage p2∆47 ... 121
Figure 25. L. lactis MG1363 proteins identified during time-course infections by phage p2 and phage p2∆47 ... 124
Figure 26. Voronoi treemaps depicting the proteotypes of L. lactis cells during phage p2∆47 infection ... 126 Figure 27. Bar graph depicting the number of bacterial proteins detected only in uninfected cultures (dark blue) or only during infection (dark red) ... 127 Figure 28. La délétion dans les BIMs résistants à p2∆47 semble former une toxine..138
Liste des tableaux
Tableau 1. Groupes de phages infectant Lactococcus lactis ... 7 Tableau 2. Information sur le génome et sur le protéome du phage p2 ... 12 Tableau 3. L. lactis MG1363 genes selected for inactivation with CRISPR-Cas9 ... 101
Liste des abréviations
ADN (DNA) Acide désoxyribonucléique (deoxyribonucleic acid)
ARN (RNA) Acide ribonucléique (ribonucleic acid)
BIM Mutant résistant aux bactériophages (bacteriophage insensitive
mutant)
BL (LAB) Bactéries lactiques (lactic acid bacteria)
Cas Associé à CRISPR (CRISPR associated)
CRISPR Regroupement de courtes régions palindromiques
régulièrement espacées (clustered regularly interspaced short
palindromic repeats)
db (ds) Double brin (double stranded)
G+C Guanine et cytosine
HNH Motif retrouvé au site catalytique des enzymes de restriction
ICTV Comité international sur la taxonomie des virus (International
Commitee on Taxonomy of Visuses)
kb Kilobases (1000 paires de bases)
kDa KiloDalton
LC-MS/MS Spectrométrie de masse en tandem
MS Spectrométrie de masse (mass spectrometry)
ORF Cadre ouvert de lecture (open reading frame)
pb (bp) Paire de bases (base pair)
RBP Protéine de liaison au récepteur (receptor binding protein)
SaV Sensibilité à AbiV
SDS-PAGE Électrophorèse sur gel de polyacrylamide sodium dodécyle
sulfate (sodium dodecyl sulfate polyacrylamide gel
electrophoresis)
SSB Protéine liant l’ADN simple-brin (Single-Stranded Binding)
SSAP Recombinase se liant à l’ADN simple-brin (Single‐Strand
Annealing Protein)
TA Toxine-antitoxine
“Life is like riding a bicycle. To keep your balance, you must keep moving.”
Remerciements
D’abord, je tiens à remercier Luc Trudel et Michel Frenette pour la confiance qu’ils ont eu en mes capacités en me référant à Sylvain Moineau. Sans eux, je n'aurais probablement pas fait d'études graduées. Preuve à l’appui, extrait d’un courriel envoyé par Sylvain le 14 janvier 2014 :
[…] je suis sur le point de commencer une période de recrutement pour au moins un(e) étudiant(e)s à la maîtrise pour l'été prochain et je voulais aller faire une petite annonce dans le cours de Physiologie bactérienne. C'est alors que Luc et Michel Frenette m'ont parlé de toi. Ils m'ont suggéré de te contacter, ce que je fais via ce courriel. Donc voilà, on ne sait pas trop ce que tu planifies à la fin de ton bacc: marché du travail, maîtrise déjà prévu avec un autre chercheur, demande en médecine, etc. Si tu es intéressée par des études à la maîtrise et que les phages pourraient peut-être t'intéresser, fais-moi signe et on pourra se rencontrer pour en discuter davantage.
La meilleure décision que j’ai prise cette année-là a été d’accepter cette offre de maîtrise avec Sylvain. Ce fut un honneur de travailler dans son laboratoire ces cinq dernières années. Je le remercie pour la grande liberté et la confiance qu’il m’a accordées. Sylvain est officier de l’ordre du Canada et un des chercheurs les plus influents au monde dans le domaine des phages. Malgré tout, il reste simple, modeste, à l’écoute, et un leader modèle. Je n’aurais pu espérer un meilleur coach scientifique.
Mon passage dans le laboratoire de Sylvain n’aurait pas été aussi agréable et enrichissant sans tous les membres de l’équipe que j’ai eu la chance de côtoyer. Bien que je ne nomme pas tout le monde ici, chacun aura agrémenté, à sa façon, mes années dans le Phage Bunker. Merci aux professionnelles de recherche : Denise Tremblay pour son savoir-faire inégalé et son rire contagieux, Geneviève Rousseau pour toutes les discussions stimulantes et sa gentillesse à la Mère Teresa, Stéphanie Loignon pour sa générosité et ses conseils. Merci à Alexia, Ariane et Moïra pour tous les fous rires, les sorties au restaurant, les activités sportives et les beaux souvenirs. Merci à Maxime Bélanger et Bruno Martel d’avoir été mes premiers mentors et amis au laboratoire. Nos discussions durant le « beer club » hebdomadaire m’ont manqué après votre départ. Je ne suis pas certaine si je devrais remercier Simon Labrie pour tous ses mauvais coups… Ton sens de l’humour et ta joie de vivre sont un atout incroyable à un milieu de travail, ne change surtout pas! Parlant de clown, le passage
de Witold Kot dans le Phage Bunker a été trop bref. Il m’a rappelé l’importance de ne pas se prendre trop au sérieux. Merci à Alexander Hynes qui agit encore à ce jour comme mentor et conseiller scientifique. Tout au long de mes études graduées, il m’a poussée afin que je développe mon plein potentiel et m’a donné des « tapes dans le dos » quand j’en avais le plus besoin.
J’ai eu la chance de développer des compétences en protéomiques en effectuant deux stages dans le domaine. Je remercie Dörte Becher de m’avoir si bien accueilli dans son laboratoire de l’Université de Greifswald en Allemagne. Merci à Andreas Otto d’avoir supervisé mes travaux et à Sandra Maaß pour avoir répondu à mes innombrables questions avec une grande patience. Je tiens également à remercier Jennifer Geddes-McAlister de l’Université de Guelph en Ontario pour m’avoir accueillie lors d’un cours stage afin que je me familiarise avec leurs protocoles d’analyses protéomiques.
Le support de mes parents a eu un impact énorme sur mes réussites universitaires. Ils ont appuyé mes décisions et par la fierté qu’ils m’ont démontrée, ils m’ont encouragé dans mon cheminement académique. C’est pourquoi cette thèse leur est dédiée. Je tiens également à remercier mon amoureux, Hakim Hoummane, qui m’appuie dans toutes les options de carrières qui s’offrent à moi, qui se montre patient lors de mes nombreuses remises en question, qui sait me rassurer et qui fait les meilleurs cafés.
J’aimerais remercier les organismes subventionnaires ayant contribué au support financier de mes recherches et de mes études graduées, soit les Fonds de recherche du Québec – Nature et technologies (FRQNT), le Conseil de recherches en sciences naturelles et en génie du Canada (CRSNG), le groupe de recherche en écologie buccale (GREB), Novalait, Op+lait et PROTEO. Enfin, merci à l’Université Laval qui offre un environnement et un milieu de vie propice au développement personnel et professionnel.
Avant-propos
Cette thèse est organisée en cinq chapitres qui incluent cinq publications, soit un chapitre de livre, un article de revue de littérature et trois articles de recherche. De plus, une publication détaillant un protocole est retrouvée en annexe.
Introduction
Les microorganismes à l’étude, soit la bactérie Lactococcus lactis subsp. cremoris MG1363 et le phage p2, sont présentés dans l’introduction de cette thèse. Les systèmes CRISPR-Cas sont d’abord abordés dans le chapitre de livre « How are genes modified? Cross breeding, mutagenesis and CRISPR-Cas9 ». Dans ce chapitre, l’origine microbienne de la technologie CRISPR-Cas9 est présentée de même que son fonctionnement et sa spécificité pour cliver l’ADN. De plus, l’édition de génome par cette technologie est détaillée. Bien que ce chapitre porte sur la modification des gènes de plantes, le mécanisme d’action est sensiblement le même pour la modification de gènes de procaryotes et de leurs virus. Les applications des systèmes CRISPR-Cas chez ces derniers sont abordés dans l’article de revue de littérature « The CRISPR-Cas app goes viral ». Enfin, la problématique, l’hypothèse et les objectifs de mes travaux de recherche sont énoncés.
How are genes modified? Cross breeding, mutagenesis and CRISPR-Cas9
Ce chapitre est sous presse pour publication dans le livre « Genetically modified and irradiated food: Controversial issues – facts versus perceptions ». Il a été édité par Veslemøy Andersen avec le groupe éditorial Elsevier.
Je suis l’auteure principale de ce chapitre. J’ai réalisé les recherches bibliographiques et j’ai rédigé le manuscrit en collaboration avec Sylvain Moineau. Au moment de la publication, Sylvain Moineau et moi-même étions affiliés au Département de biochimie, de microbiologie et de bio-informatique, au Centre de référence pour virus bactériens Félix d'Hérelle de l'Université Laval et au Groupe de recherche en écologie buccale.
Les numéros des figures et le format des références ont été modifiés pour être conforme avec la thèse.
The CRISPR-Cas app goes viral
Cet article a été publié le 14 juin 2017 dans le 37e volume du journal Current Opinion in
Microbiology aux pages 103-109.
Je suis l’auteure principale de cet article. J’ai réalisé les recherches bibliographiques et j’ai rédigé le manuscrit en collaboration avec Sylvain Moineau et Philippe Horvath. Au moment de la publication, Sylvain Moineau et moi-même étions affiliés au Département de biochimie, de microbiologie et de bio-informatique, au Centre de référence pour virus bactériens Félix d'Hérelle de l'Université Laval et au Groupe de recherche en écologie buccale. Philipe Horvath était affilié à DuPont Nutrition and Health à Dangé-Saint-Romain en France.
Les numéros des figures et le format des références ont été modifiés pour être conforme avec la thèse.
Premier article de recherche (chapitre 1)
Genome engineering of lactococcal phages using CRISPR-Cas9
Cet article a été publié en ligne le 21 mars 2017 et imprimé le 21 juillet 2017 dans le 6e
volume du journal ACS Synthetic Biology aux pages 1351-1358.
Je suis l'auteure principale de cet article. J'ai participé à l'élaboration de l'étude, réalisé l'ensemble des expériences et des analyses et rédigé le manuscrit. Denise M. Tremblay a séquencé le génome des phages mutants. Sylvain Moineau a participé à l'élaboration de l'étude, supervisé l'ensemble des travaux et révisé le manuscrit. Au moment de la publication, tous les auteurs étaient affiliés au Département de biochimie, de microbiologie et de bio-informatique, au Centre de référence pour virus bactériens Félix d'Hérelle de l'Université Laval et au Groupe de recherche en écologie buccale.
Les numéros des figures et le format des références ont été modifiés pour être conforme avec la thèse. Le matériel supplémentaire est disponible en ligne (doi: 10.1021/acssynbio.6b00388).
Deuxième article de recherche (chapitre 2)
Investigating Lactococcus lactis MG1363 response to phage p2 infection at the proteome level
Cet article a été publié en ligne le 24 janvier 2019 et imprimé le 1er avril 2019 dans le 18e
volume du journal Molecular and Cellular Proteomics aux pages 704-714.
Je suis l'auteure principale de cet article. J'ai participé à l'élaboration de l'étude, réalisé la majeure partie des expériences et des analyses et j’ai rédigé le manuscrit. Andreas Otto a participé à l’acquisition et à l’analyse des données de spectrométrie de masse. Sandra Maaß a contribué à l'analyse de ces mêmes données et à la rédaction du manuscrit. Kristina Plate a réalisé les outils de visualisation Voronois. Dörte Becher a participé à l'élaboration de l'étude et a supervisé une partie des travaux. Sylvain Moineau a participé à l'élaboration de l'étude, supervisé une partie des travaux et révisé le manuscrit. Au moment de la publication, Sylvain Moineau et moi-même étions affiliés au Département de biochimie, de microbiologie et de bio-informatique, au Centre de référence pour virus bactériens Félix d'Hérelle de l'Université Laval et au Groupe de recherche en écologie buccale. Andreas Otto, Sandra Maaß, Kristina Plate et Dörte Becher étaient affiliés à l'Institut de microbiologie de l'Université de Greifswald en Allemagne.
Les numéros des figures et le format des références ont été modifiés pour être conforme avec la thèse. Le matériel supplémentaire est disponible en ligne (doi: 10.1074/mcp.RA118.001135).
Troisième article de recherche (chapitre 3)
A phage protein impedes Lactococcus lactis resistance to phage infection
Cet article est en préparation pour soumission.
Je suis l'auteure principale de cet article. J'ai participé à l'élaboration de l'étude, réalisé la majeure partie des expériences et des analyses et rédigé le manuscrit. Sandra Maaß et Andreas Otto ont contribué à l’acquisition et à l'analyse des données de spectrométrie de masse. Jérémie Hamel a réalisé les travaux de résonance magnétique. Pier-Luc Plante a
contribué aux analyses génomiques. Geneviève M. Rousseau a fait les tests d'adsorption. Denise M. Tremblay a contribué à l’obtention des résultats d’analyses génomiques. Rong Shi a contribué à la caractérisation de la structure de l’ORF47. Stéphane M. Gagné a supervisé les travaux de résonance magnétique et effectué les recherches d’homologie de séquence et de structure. Jacques Corbeil a supervisé une partie des travaux d’analyses génomiques. Dörte Becher a participé à l'élaboration de l'étude et a supervisé les travaux de spectrométrie de masse. Sylvain Moineau a participé à l'élaboration de l'étude, supervisé une partie des travaux et révisé le manuscrit. Au moment de la publication, Sylvain Moineau, Geneviève M. Rousseau, Denise M. Tremblay et moi-même étions affiliés au Département de biochimie, de microbiologie et de bio-informatique, à PROTEO, au Centre de référence pour virus bactériens Félix d'Hérelle de l'Université Laval et au Groupe de recherche en écologie buccale. Jérémie Hamel, Rong Shi et Stéphane M. Gagné étaient affiliés au Département de biochimie, de microbiologie et de bio-informatique, à PROTEO et à l'Institut de biologie intégrative et des systèmes. Pier-Luc Plante et Jacques Corbeil étaient affiliés au Centre de recherche en infectiologie de l'Université Laval, au Centre de recherche du CHU de Québec, au Centre de recherche en données massives et au Département de médecine moléculaire. Andreas Otto, Sandra Maaß et Dörte Becher étaient affiliés à l'Institut de microbiologie de l'Université de Greifswald en Allemagne.
Introduction
Lactococcus lactis
Caractéristiques principales
Lactococcus lactis est l’une des premières espèces bactériennes domestiquées par l’homme,
soit pour la production de fromages. Bien que son identité fût alors inconnue, on savait qu’en inoculant du lait frais avec un échantillon d’une fermentation passée, on provoquait une acidification du lait, ce qui apportait des avantages en termes de conservation, texture, arômes et saveurs. C’est à la fin du XIXe siècle, après la publication des travaux de Pasteur sur les
fermentations lactiques, que Joseph Lister obtint la première culture pure de L. lactis, qu’il baptisa à l’époque Bacterium lactis (Lister, 1873). Avant d’être nommée L. lactis, cette espèce fût également connue sous le nom de Streptococcus lactis. Ce n’est qu’en 1985 que le genre Lactococcus de même que les sous-espèces L. lactis ssp. cremoris et L. lactis ssp.
lactis ont été proposés (Schleifer et al., 1985). Quelques autres espèces de lactocoques, dont L. piscium et L. garvieae, qui ne sont pas exploitées pour la fermentation du lait, mais qui
sont plutôt associées à la détérioration des aliments, à la mastite bovine ou à la lactococcose chez les poissons d’élevage ont également été identifiées et caractérisées (Pothakos et al., 2014; Rahkila et al., 2012; Teuber and Geis, 2006).
L. lactis est une espèce mésophile régulièrement consommée à travers le monde et elle
contribue à la santé et au bien-être de millions de gens. En plus de son utilisation dans les fermentations alimentaires, L. lactis est exploitée pour une multitude d’applications biotechnologiques, ce qui en fait une des espèces bactériennes les plus importantes pour l’économie mondiale. Il s’agit d’ailleurs du premier organisme génétiquement modifié utilisé comme véhicule de livraison de protéines pour le traitement d’une maladie humaine, soit la maladie de Crohn (Braat et al., 2006). L. lactis est également étudiée comme vecteurs de vaccins oraux et pour l’expression de protéines et de peptides antimicrobiens, dont une variété de lantibiotiques (Mierau and Kleerebezem, 2005; Wyszynska et al., 2015). De plus, sa nature sécuritaire (Generally Recognized As Safe; GRAS) en fait une alternative à
recombinantes (Morello et al., 2008). Ces applications n’auraient pas été possibles sans les connaissances acquises sur la biologie de L. lactis au cours des 50 dernières années (Kok et al., 2017). En effet, de nombreux travaux d’enzymologie, de biochimie et de physiologie ont contribués à la caractérisation de cet organisme devenu un modèle pour l’étude des bactéries à Gram positif.
L. lactis fait partie du regroupement fonctionnel des bactéries lactiques (BL). Les BL
comprennent des genres bactériens à Gram positif, aérotolérantes et produisant de l’acide lactique à partir de la fermentation des hexoses (Makarova et al., 2006). Selon les produits finaux de fermentations, les BL sont classées en deux groupes, soit les homolactiques et les hétérolactiques. Alors que les bactéries homolactiques produisent uniquement de l’acide lactique à partir de la fermentation des sucres, les bactéries hétérolactiques produisent également de l’éthanol, du dioxyde de carbone et des produits comme l’acétaldéhyde et le diacétyle qui procurent arômes et flaveurs (Endo and Dicks, 2014; Kandler, 1983). L. lactis est une bactérie homolactique et se présente sous forme de diplocoques ou de coques en courtes chaînettes dont les unités ont un diamètre de 0,5 à 1,5 µm. La sous-espèce cremoris est utilisée pour la production de certains fromages. Par exemple, elle compose jusqu’à 98% des cultures de départ pour la production de cheddar et de camembert (Teuber and Geis, 2006).
Importance de la souche MG1363
La souche L. lactis ssp. cremoris MG1363 (numéro d’accession GenBank AM406671) est le prototype international pour la génétique des BL et un modèle pour l’étude des bactéries à Gram positif. Dépourvue de plasmide et de prophages, elle est dérivée de la souche NCDO712 retrouvée dans les cultures de départ utilisées pour la transformation des produits laitiers (Gasson, 1983). Puisque les gènes nécessaires au métabolisme du lactose sont plasmidiques chez les lactocoques (McKay, 1983), la souche MG1363 ne peut utiliser ce sucre comme source d’énergie et nécessite un apport de glucose. L’entièreté du génome de
L. lactis MG1363 se retrouve sur un chromosome de 2,53 Mbp dont la séquence complète
est connue. Le contenu en guanine et cytosine (G+C) est faible avec un pourcentage s’élevant à 35,7%. Il contient 81 pseudogènes et encode 2437 protéines (Linares et al., 2010; Silva et al., 2018; Wegmann et al., 2007).
Les souches industrielles de L. lactis contiennent généralement plusieurs plasmides de tailles allant de 2 à 100 kb (Klaenhammer et al., 1978). En plus du métabolisme du lactose, ces plasmides peuvent coder pour des gènes de fermentation du citrate, de production d’exopolysaccharide, de protéolyse et de résistance aux bactériophages (Ainsworth et al., 2014). Nombre de ces plasmides, surtout les plus petits, ont été utilisés pour construire des vecteurs de clonage. Un grand avantage de ces vecteurs plasmidiques est leur large spectre. En effet, certains peuvent se répliquer dans une grande variété de BL, dans plusieurs genres bactériens à Gram positif, et même dans le modèle bactérien le plus étudié, E. coli (Kok et al., 1984; Vosman and Venema, 1983). La disponibilité de ces vecteurs de clonage et la découverte de la technique d’électroporation (Harlander, 1987) ont propulsé l’étude de L.
lactis dans l’ère du génie génétique.
Les phages
Les bactériophages, ou plus simplement les phages, sont des parasites obligatoires qui infectent spécifiquement les bactéries. Il s’agit de l’entité biologique la plus abondante et la plus diversifiée sur Terre avec une population globale estimée à 1031 (Breitbart and Rohwer,
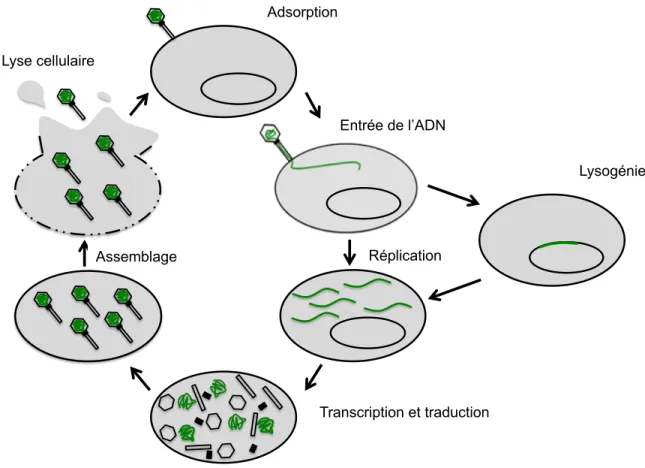
2005; Suttle, 2005). On les retrouve dans les mêmes niches écologiques que leurs hôtes où ils assurent l’équilibre des écosystèmes par le contrôle des populations bactériennes (Fuhrman, 1999). Comme tout parasite obligatoire, les phages ont besoin d’une cellule hôte pour se répliquer. Lors d’une infection par un phage virulent, la multiplication des particules virales se fait selon un cycle dit lytique (Figure 1).
Le cycle de multiplication
La première étape du cycle consiste en l’adsorption, soit l’attachement spécifique du phage à un récepteur se situant à la surface d’une cellule bactérienne via une protéine de liaison au récepteur (Receptor Binding Protein; RBP). Les peptidoglycanes, les acides téichoïques et lipotéichoïque et les protéines associées à la paroi sont des récepteurs potentiels pour l’attachement des phages à la surface des bactéries à Gram positif (Kutter et al., 2005). Le génome des phages peut être constitué d’une macromolécule d’acide désoxyribonucléique (ADN) ou d’acide ribonucléique (ARN). La quasi-totalité des phages isolés à ce jour ont un génome constitué d’ADN double brin (ADNdb) (Ackermann and Prangishvili, 2012;
Maniloff and Ackermann, 1998). Après l’adsorption, l’ADN linéaire de ces phages est éjecté de la capside et pénètre dans la cellule cible en passant par la queue. Une fois dans la cellule, l’ADN viral peut s’intégrer dans le chromosome bactérien sous forme de prophage, Dans cette situation, on parle de lysogénie et on décrit la cellule comme étant lysogène. Lors de la division cellulaire, chaque cellule fille héritera de ce prophage. Éventuellement, l’ADN viral sera excisé du chromosome et le cycle lytique reprendra.
Certains phages ne peuvent pas effectuer cette étape de lysogénie, et leur ADN est circularisé et répliqué dès l’entrée dans la bactérie. La réplication se fait selon un mode de cercle roulant ou un mode thêta. Dans les deux cas, de longs concatémères de plusieurs génomes de phages sont produits (Weigel and Seitz, 2006). La réplication de l’ADN viral se déroule à des sites intracellulaires spécifiques. Cette localisation de la réplication est possible grâce à l’action de protéines du cytosquelette (Erb and Pogliano, 2013). Les génomes de phages sont souvent organisés en modules dans lesquels sont regroupés des gènes aux fonctions similaires (Botstein, 1980; Veesler and Cambillau, 2011). Les gènes sont transcrits d’une manière hautement régulée et sont classés selon le moment de leur expression lors de l’infection. D’abord, les gènes précoces sont exprimés. Ceux-ci codent généralement pour des protéines non structurales impliquées dans la réplication de l’ADN viral et le contrôle de la machinerie cellulaire de l’hôte. Ensuite, les gènes médians sont transcrits. Bien que leurs fonctions soient souvent inconnues, certaines protéines médianes semblent importantes pour la régulation du cycle lytique. Enfin, les gènes tardifs codent pour des protéines impliquées dans la morphogenèse des virions, l’encapsidation de l’ADN et la lyse de la cellule hôte. La production des protéines tardives et l’auto-assemblage de certaines d’entre elles mènent ensuite à l’encapsidation de génomes nouvellement synthétisés. Lorsque les particules virales sont assemblées et que l’environnement est adéquat, la holine s’accumule dans la cellule et forme des pores dans la membrane pour permettre à l’endolysine de dégrader le peptidoglycane de la paroi et causer la lyse de la cellule. De nouveaux phages sont ainsi relâchés et chacun a le potentiel d’infecter une nouvelle bactérie afin de recommencer le cycle lytique.
Figure 1. Schématisation du cycle de multiplication des phages virulents Les protéines non structurales
De manière générale, les protéines non structurales de phages sont moins conservées et plus difficiles à étudier que les protéines structurales puisqu’elles ne font pas partie intégrante des particules virales. Elles sont plutôt retrouvées à l’intérieur des cellules hôtes ou relarguéee dans l’environnement lors du cycle de réplication. Malgré l’expansion continuelle des bases de données et le développement des algorithmes de recherche, il est rare de trouver des similarités de séquences entre les protéines phagiques non structurales (Altschul et al., 1990; Kelley and Sternberg, 2009).
La plupart des protéines phagiques non structurales sont de petits polypeptides synthétisés dès le début de l’infection et servent à inhiber, activer ou rediriger divers processus intracellulaires de manière à favoriser la production de particules virales (Roucourt and Lavigne, 2009). Généralement, ces protéines ont des propriétés bactériostatiques et sont non essentielles pour la réplication des phages en conditions de laboratoire (Liu et al., 2004;
Adsorption
DNA entry
Replication
Transcription and translation Assembly Lysis Lysogeny Entrée de l’ADN Adsorption Lysogénie Réplication Transcription et traduction Lyse cellulaire Assemblage
Roach and Donovan, 2015; Yano and Rothman-Denes, 2011). Il est probable qu’elles jouent un rôle important dans leur adaptation à une niche écologique particulière et à la fluctuation des conditions physiologiques (Koskella and Brockhurst, 2014). Bien que l’élucidation de la fonction de ces petites protéines phagiques soit prometteuse pour diverses applications thérapeutiques et pour la compréhension des infections virales en général, plusieurs d’entre elles demeurent sans fonction connue (Wagemans and Lavigne, 2012). Elles sont plus difficiles à caractériser que les protéines structurales par des techniques de microscopie électronique, de SDS-PAGE et de spectrométrie de masse (Fokine and Rossmann, 2014). L’élucidation de leur fonction repose surtout sur l’analyse de l’impact de leur inactivation in
vivo dans le cycle de multiplication du phage. Les inactivations de gènes in vivo sont d’autant
plus difficiles pour les phages strictement lytiques puisque leur génome n’est que de court passage dans la cellule bactérienne. Or, l’édition du génome doit habituellement se faire au cours d’un cycle infectieux.
Les phages dans l’industrie laitière
Les phages sont naturellement présents dans le lait et certains résistent à la pasteurisation. Ils représentent donc un risque d’infection des souches bactériennes soigneusement sélectionnées et utilisées pour la transformation de divers produits laitiers. Des systèmes industriels de contrôle de ces parasites ont été développés, mais leur optimisation est toujours d’actualité. Selon l’usine, les phages peuvent affecter négativement jusqu’à 10% des fermentations laitières (Moineau and Lévesque, 2005). Près de 200 phages de lactocoques ont été isolés dans des usines québécoises. Parmi ceux-ci, 90% appartient au genre
Skunavirus (auparavant 936), clairement le plus problématique dans l’industrie fromagère
(Mahony et al., 2012; Oliveira et al., 2018; Sadiq et al., 2018).
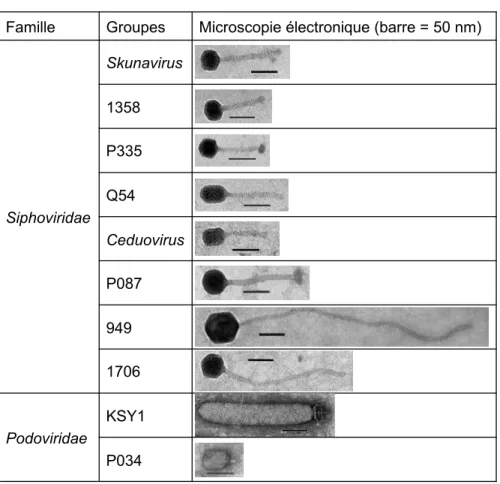
Tous les phages de lactocoques isolés jusqu’à présent font partie de l’ordre des Caudovirales. Selon leurs caractéristiques morphologiques et génomiques, les phages sont classifiés par le Comité international de taxonomie des virus (International Commitee on Taxonomy of
Visuses; ICTV). Les Caudovirales possèdent tous une queue et leur matériel génétique se
présente sous forme d’ADNdb contenu dans une capside icosaédrique. Selon la morphologie de leur queue, les Caudovirales sont présentement classifiés en cinq familles. Les
alors que les Siphoviridae ont une longue queue non contractile et les Podoviridae ont une courte queue non contractile (Ackermann, 2007). Ces familles regroupent plus de 96% des phages isolés à ce jour (Ackermann and Prangishvili, 2012; Maniloff and Ackermann, 1998). Les phages infectant L. lactis sont majoritairement des Siphoviridae. On les classe en dix groupes, dont les Skunavirus (Chandry et al., 1997), Ceduovirus (Jarvis et al., 1994) et P335 (Labrie et al., 2008) sont, en ordre d’importance, les plus problématiques pour l’industrie laitière, et ce à l’échelle internationale (Deveau et al., 2006; Mahony et al., 2012; Rousseau and Moineau, 2009) (Tableau 1). Les genres Skunavirus et Ceduovirus comportent uniquement des phages virulents, donc incapables d’insérer leur génome dans le chromosome bactérien sous forme de prophage, alors que le groupe P335 comporte également des phages tempérés, capables d’une telle lysogénie (Deveau et al., 2006; Madsen et al., 2001).
Tableau 1. Groupes de phages infectant Lactococcus lactis
Modifié de (Deveau et al., 2006).
Famille Groupes Microscopie électronique (barre = 50 nm)
Siphoviridae Skunavirus 1358 P335 Q54 Ceduovirus P087 949 1706 Podoviridae KSY1 P034
Lors de la production de fromages, des cultures bactériennes définies ou indéfinies sont inoculées dans le lait. Les cultures indéfinies contiennent un nombre inconnu de multiples souches de BL dérivées de pratiques traditionnelles et sélectionnées pour les qualités organoleptiques qu’elles procurent au produit final. Elles sont surtout utilisées dans les productions artisanales. Aujourd’hui, les cultures de départ définies sont plus fréquentes puisqu’elles sont mieux caractérisées, elles ont été optimisées pour des propriétés fermentaires spécifiques et elles sont plus faciles à contrôler. Comme leur nom l’indique, elles sont composées d’un mélange défini de cultures pures. Puisque la biodiversité retrouvée dans les cultures définies est moindre que dans les cultures indéfinies, les conséquences d’une infection phagique y sont généralement plus désastreuses (Auclair and Accolas, 1983; Daly, 1983). Les cultures définies sont utilisées en rotation selon leur profil de sensibilité aux phages afin de limiter ce problème (Sing and Klaenhammer, 1993). Malgré la rotation des souches et les mesures sanitaires prises en usine, la présence de phages demeure une menace bien présente. Les phages virulents de BL les plus efficaces complètent le cycle lytique en moins de 30 minutes et plus d’une centaine de phages peuvent être relâchés par cellule infectée (Samson and Moineau, 2013). L’optimisation des stratégies de défense est toujours d’actualité et celles-ci doivent être adaptées selon l’usine et la population de phages qui l’affecte (Mercanti et al., 2012; Vodzinska et al., 2011). Une meilleure connaissance de la biologie des phages infectant les lactocoques permettra certainement un meilleur contrôle de ce parasite industriel.
Le phage modèle p2
Le phage p2 (numéro d’accession GenBank GQ979703) est un virus modèle du genre
Skunavirus. Son génome est constitué de 27 595 bp et de 49 cadres ouverts de lecture (open reading frames; orfs) qui ont été prédits par des analyses bioinformatiques. Comme pour son
hôte L. lactis MG1363, le contenu en G+C du génome de p2 est faible avec un pourcentage s’élevant à 34,7%. Il fait partie des phages les plus étudiés à travers le monde et toutes ses protéines structurales sont connues (Bebeacua et al., 2013). Les virions de p2 ont une longueur approximative de 210 nm. Ils sont constitués d’une capside de 69 nm attachée par un connecteur à une longue queue non contractile dont l’extrémité est dotée d’un complexe protéique appelé plaque basale (Figure 2).
Figure 2. Représentation du génome de p2
Les gènes sont représentés par des flèches et les couleurs de ces dernières indiquent l’ordre de leur expression lors de l’infection. Les gènes tardifs (blancs) codent pour des protéines structurales déjà bien caractérisées alors que les gènes précoces et médians (vert et jaune, respectivement), codent pour des protéines aux fonctions encore inconnues. La structure du phage p2 a été modifiée de (Bebeacua et al., 2013).
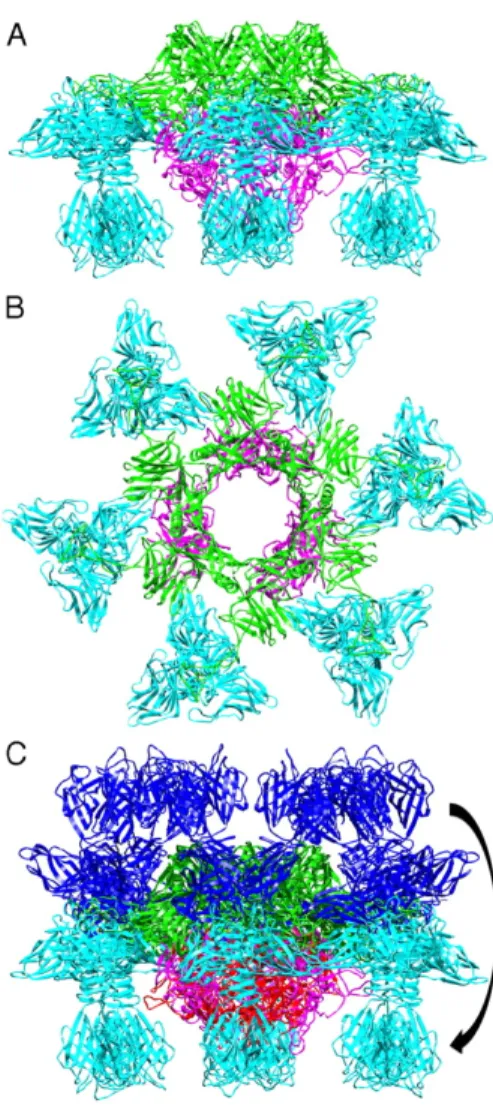
La plaque basale de p2 permet la reconnaissance d’un récepteur de nature polysaccharidique à la surface de certaines souches de L. lactis. Elle est constituée d’un hexamère d’ORF15, d’un trimère d’ORF16 et de six trimères d’ORF18 (RBP). En présence de calcium, la plaque basale subit un important changement de conformation qui permet d’orienter correctement ses RBPs vers les récepteurs se retrouvant à la surface de la bactérie hôte, d’ouvrir un canal dans la plaque basale pour permettre la sortie de l’ADN viral et ainsi initier l’infection (Sciara et al., 2010) (Figure 3).
Tardifs Précoces Médians
?
Late Earl y Mid dle 0.0 0.4 0.8 T0 T10 T20 T40 1 2 3 4 5 6 7 8 1011 1213 1415 1617 1819 2022 2324 2526 2932 3435 3637 3839 4041 4243 4445 48 1 2 3 4 5 6 7 9 8 10 11 12 13 14 15 16 1718 19 20 21 22 23 24 25 26 28 29 3132 33 3435 36 37 38 39 40 41 42 43 44 45 46 47 48 49 30 27 1 2 3 4 5 6 7 9 8 10 11 12 13 14 15 16 1718 19 20 21 22 23 24 25 26 28 29 3132 33 3435 36 37 38 39 40 41 42 43 44 45 46 47 48 49 30 27?
1 2 3 4 5 6 7 98 10 11 1213 14 15 16 17 18 19 20212223242526 28 29 31 323334 35363738394041 42 43 44 4546 47 4849 27 30Figure 3. Changement de conformation de la plaque basale de p2 en présence de calcium
Les ORF15, ORF16 et ORF18 sont schématisés en vert, rose et bleu, respectivement. La structure de la plaque basale est représentée avec une vue de côté (A) et une vue d’en haut (B). En (C), les trimères d’ORF18 ont subi une rotation de 200°.
Les étapes d’assemblage des virions sont peu connues chez les phages de lactocoques. Néanmoins, il a été suggéré que l’ORF12, une protéine tardive et non structurale, agirait comme chaperone s’associant à la région hydrophobique de la protéine d’échafaudage afin de maintenir cette dernière en solution avant l’assemblage de la protéine majeure de la queue (Siponen et al., 2009). Les protéines d’échafaudage servent de gabarit pour la mesure de la queue lors de l’assemblage (Katsura, 1987). Bien que peu de données soient disponibles sur l’encapsidation de l’ADN et la formation des capsides, il a été proposé que l’ORF6 de p2, la protéine majeure de la capside, est clivée par une protéase (ORF5) et forme des procapsides
l’endolysine (ORF20) vont lyser la cellule pour relâcher la progéniture. La holine va former un pore dans la membrane bactérienne pour permettre à l’endolysine qui s’accumule dans la cellule d’avoir accès à la paroi (Catalao et al., 2013). Lorsque les domaines de type amidase ou de protéines dégradant le peptidoglycane entrent en contact avec la paroi, celle-ci est dégradée, ce qui mène à l’éclatement des bactéries et le relargage de particules virales matures.
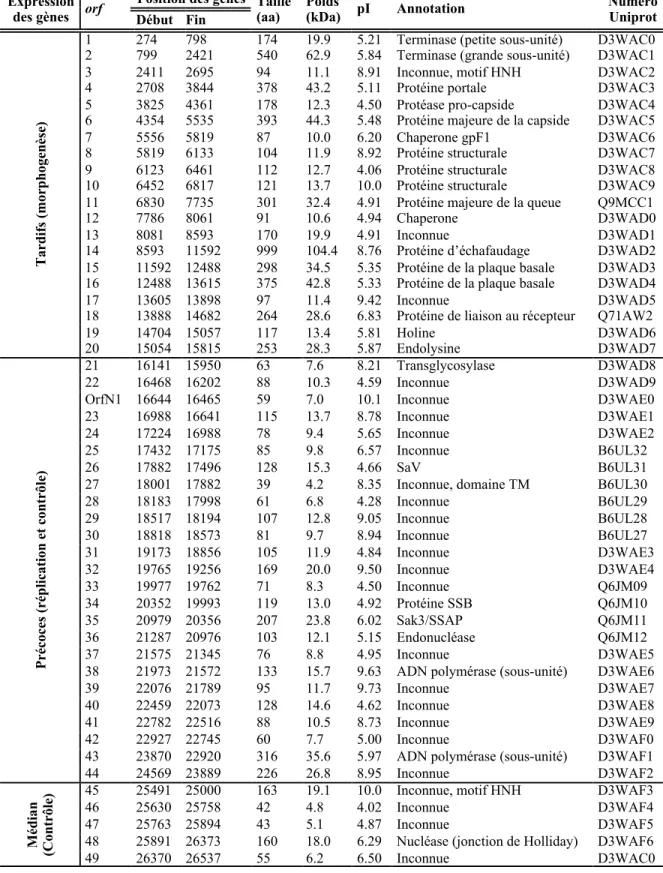
Le Tableau 2 contient des informations pertinentes sur l’état actuel des connaissances sur le protéome du phage p2. À l’exception de quelques orfs, la plupart des gènes précoces et médians codent pour des protéines aux fonctions encore inconnues. Une de ces exceptions est le gène précoce orf26 qui code pour la protéine SaV impliquée dans la sensibilité au mécanisme bactérien d’avortement de l’infection AbiV (Abi; Abortive infection) (Haaber et al., 2009). Il a été démontré que la protéine bactérienne AbiV interagit directement avec SaV, et prévient ainsi la traduction des autres protéines de phages (Haaber et al., 2010). Les protéines non structurales encodées par les gènes précoces orf34 et orf35 du phage p2 ont également été caractérisées. L’ORF34 est une protéine liant l’ADN simple-brin (SSB;
Single-Stranded Binding Protein) (Scaltriti et al., 2009). Pour sa part, l’ORF35, aussi appelée Sak3,
est une ATPase (Scaltriti et al., 2011) impliquée dans la sensibilité au mécanisme de résistance AbiV (Bouchard and Moineau, 2004). Cette dernière fait également partie du groupe des recombinases SSAP (Single‐Strand Annealing Protein), ces protéines se lient à l’ADN simple-brin et favorisent l’assemblage de brins complémentaires. L’hypothèse actuel est que SSB recruterait Sak3 pour stimuler des événements de recombinaison homologue (Scaltriti et al., 2011). L’élucidation de la fonction de ces protéines non structurales ont été possibles grâce à l’analyse de phages mutants obtenus naturellement en laboratoire et par des analyses biochimiques in vitro.
Jusqu’à tout récemment, l’absence d’un outil efficace pour inactiver ou modifier les gènes de phages virulents était un important facteur limitant pour les études in vivo des protéines de phages non structurales.
Tableau 2. Information sur le génome et sur le protéome du phage p2
Expression
des gènes orf Début Fin Position des gènes Taille (aa) Poids (kDa) pI Annotation Numéro Uniprot
Tar d ifs (mor p h oge n ès e)
1 274 798 174 19.9 5.21 Terminase (petite sous-unité) D3WAC0
2 799 2421 540 62.9 5.84 Terminase (grande sous-unité) D3WAC1
3 2411 2695 94 11.1 8.91 Inconnue, motif HNH D3WAC2
4 2708 3844 378 43.2 5.11 Protéine portale D3WAC3
5 3825 4361 178 12.3 4.50 Protéase pro-capside D3WAC4
6 4354 5535 393 44.3 5.48 Protéine majeure de la capside D3WAC5
7 5556 5819 87 10.0 6.20 Chaperone gpF1 D3WAC6
8 5819 6133 104 11.9 8.92 Protéine structurale D3WAC7
9 6123 6461 112 12.7 4.06 Protéine structurale D3WAC8
10 6452 6817 121 13.7 10.0 Protéine structurale D3WAC9
11 6830 7735 301 32.4 4.91 Protéine majeure de la queue Q9MCC1
12 7786 8061 91 10.6 4.94 Chaperone D3WAD0
13 8081 8593 170 19.9 4.91 Inconnue D3WAD1
14 8593 11592 999 104.4 8.76 Protéine d’échafaudage D3WAD2
15 11592 12488 298 34.5 5.35 Protéine de la plaque basale D3WAD3
16 12488 13615 375 42.8 5.33 Protéine de la plaque basale D3WAD4
17 13605 13898 97 11.4 9.42 Inconnue D3WAD5
18 13888 14682 264 28.6 6.83 Protéine de liaison au récepteur Q71AW2
19 14704 15057 117 13.4 5.81 Holine D3WAD6 20 15054 15815 253 28.3 5.87 Endolysine D3WAD7 P ré coc es (r ép li cati on e t c on tr ôl e) 21 16141 15950 63 7.6 8.21 Transglycosylase D3WAD8 22 16468 16202 88 10.3 4.59 Inconnue D3WAD9
OrfN1 16644 16465 59 7.0 10.1 Inconnue D3WAE0
23 16988 16641 115 13.7 8.78 Inconnue D3WAE1
24 17224 16988 78 9.4 5.65 Inconnue D3WAE2
25 17432 17175 85 9.8 6.57 Inconnue B6UL32
26 17882 17496 128 15.3 4.66 SaV B6UL31
27 18001 17882 39 4.2 8.35 Inconnue, domaine TM B6UL30
28 18183 17998 61 6.8 4.28 Inconnue B6UL29 29 18517 18194 107 12.8 9.05 Inconnue B6UL28 30 18818 18573 81 9.7 8.94 Inconnue B6UL27 31 19173 18856 105 11.9 4.84 Inconnue D3WAE3 32 19765 19256 169 20.0 9.50 Inconnue D3WAE4 33 19977 19762 71 8.3 4.50 Inconnue Q6JM09 34 20352 19993 119 13.0 4.92 Protéine SSB Q6JM10 35 20979 20356 207 23.8 6.02 Sak3/SSAP Q6JM11 36 21287 20976 103 12.1 5.15 Endonucléase Q6JM12 37 21575 21345 76 8.8 4.95 Inconnue D3WAE5
38 21973 21572 133 15.7 9.63 ADN polymérase (sous-unité) D3WAE6
39 22076 21789 95 11.7 9.73 Inconnue D3WAE7
40 22459 22073 128 14.6 4.62 Inconnue D3WAE8
41 22782 22516 88 10.5 8.73 Inconnue D3WAE9
42 22927 22745 60 7.7 5.00 Inconnue D3WAF0
43 23870 22920 316 35.6 5.97 ADN polymérase (sous-unité) D3WAF1
44 24569 23889 226 26.8 8.95 Inconnue D3WAF2 M éd ian (C on tr ôl
e) 45 46 25491 25000 25630 25758 163 42 19.1 4.8 10.0 Inconnue, motif HNH 4.02 Inconnue D3WAF3 D3WAF4
47 25763 25894 43 5.1 4.87 Inconnue D3WAF5
48 25891 26373 160 18.0 6.29 Nucléase (jonction de Holliday) D3WAF6
Genetically modified and irradiated food: Controversial issues – facts
versus perceptions
Chapter 4: How are genes modified? Cross breeding, mutagenesis and CRISPR-Cas9
Marie-Laurence Lemay1,2 and Sylvain Moineau1,2,3 *
1 Département de biochimie, de microbiologie, et de bio-informatique, Faculté des sciences
et de génie, Université Laval, Québec City, QC, Canada
2 Groupe de recherche en écologie buccale, Faculté de médecine dentaire, Université Laval,
Québec City, QC, Canada
3 Félix d'Hérelle Reference Center for Bacterial Viruses, Faculté de médecine dentaire,
Résumé
Les humains peuvent maintenant déchiffrer les lettres du code génétique à un rythme impressionnant. Les gènes, définis comme des séquences d’ADN spécifiques, agissent comme des instructions déterminant les caractéristiques de tous les organismes et de leurs virus. La découverte et l’adaptation de ciseaux moléculaires précis ont généré des possibilités pour modifier des gènes avec une précision auparavant inégalée. Pour ce faire, les ciseaux sont d’abord équipés d’un dispositif de repérage afin de cibler une région particulière d’un génome. Ensuite, ils sont introduits dans le noyau d’une cellule où ils induisent un bris d’ADN précis dans la région cible. Le dommage à l’ADN est ensuite réparé par la machinerie cellulaire, ce qui peut mener à une mutation voulue affectant un trait d’intérêt chez un organisme. Cette approche dirigée de l’édition de génome a mené à d’innombrables découvertes dans tous les domaines de la biologie, incluant les sciences agricoles. L’outil recevant le plus d’attention dans cette révolution génétique est CRISPR-Cas9.
Abstract
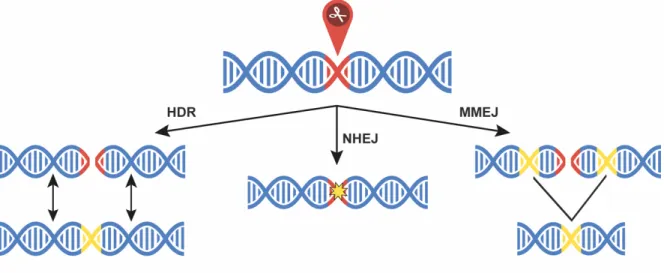
Humans can now decipher the letters of the DNA alphabet at a breathtaking pace. Genes, defined as specific DNA sequences, act as instructions that determine the characteristics of all cells and their viruses. The discovery and adaptation of precise molecular scissors has created new possibilities to modify genes at an unprecedented precision. In order to do so, the scissors are first equipped with a tracking device to target a well-defined region within the vast genome. Next, they are introduced into the cell nucleus where they induce a precise DNA break in the targeted gene or genome area. The DNA damage is then repaired by the cellular machinery that can lead to an intended mutation affecting the desired trait of an organism. This directed approach of genome editing has led to countless breakthroughs in applications across biology, including agricultural sciences. The tool receiving the most attention in this genome editing revolution is CRISPR-Cas9.
Glossary
5’-NRG PAM recognized by SpCas9, where N = A, C, G or T and R = A or G 5’-TTN PAM recognized by FnCpf1, where N = A, C, G or T
A adenine
AAV adeno-associated viral vector C cytosine
Cas gene gene coding for Cas protein Cas protein CRISPR associated protein
Cas9 a specific signature Cas protein, a nuclease used for genome editing CjCas9 Cas9 of Campylobacter jejuni
Cpf1 CRISPR-associated nuclease in Prevotella and Francisella CRISPR clustered regularly interspaced short palindromic repeats CRISPR-Cas9 gene-editing tool using Cas9
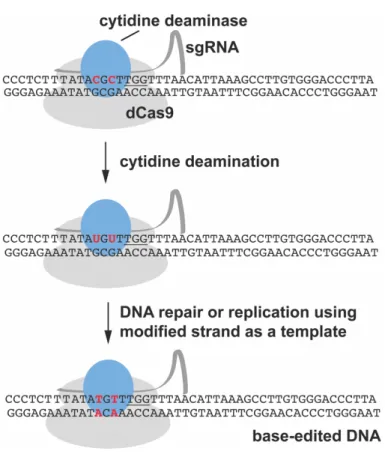
crRNA CRISPR RNA dictating the specificity of the CRISPR-Cas9 tool dCas9 catalytically dead Cas9
DNA deoxyribonucleic acid DSB double-strand break
FnCpf1 Cpf1 from Francisella novicida
G guanine
GMO genetically modified organism HDR homology-directed recombination HGT horizontal gene transfer
HNH Cas9 nuclease domain cleaving the strand complementary to the spacer MMEJ microhomology-mediated end joining
nCas9 Cas9 nickase cleaving only one strand of the DNA double helix NHEJ non-homologous end joining
NmCas9 Cas9 of Neisseria meningitidis Nuclease enzyme acting as molecular scissor
PAM protospacer adjacent motif (2-6 base pair DNA sequence) PEG polyethylene glycol
phages bacterial viruses RNA ribonucleic acid RNP ribonucleoprotein
RuvC Cas9 nuclease domain cleaving the strand non-complementary to the spacer SaCas9 Cas9 of Staphylococcus aureus
sgRNA single guide RNA, combination of crRNA and the tracrRNA SpCas9 Cas9 of Streptococcus pyogenes
T thymine
T‐DNA transferred DNA
TALEN transcription activator-like effector nuclease Ti tumour‐inducing
tracrRNA trans-activating CRISPR RNA ZFN zinc finger nuclease
Introduction
At the beginning of agriculture, farmers promptly started to domesticate plants to produce crops with more desirable characteristics than their wild-type counterparts. Without any theoretical knowledge of genetics, they crossbred plants with different traits to produce improved hybrids. It was only in the mid-19th century that Gregor Mendel described his
theory of heredity using a series of pea plant hybridization experiments (Mendel, 1866). His laws of inheritance explained how traits (phenotypes) were passed on from parents to offspring in a non-random fashion through defined elements (genes). It took over three decades for the scientific community (and the general public) to acknowledge his work, thus hindering advancements in modern genetics. In the 20th century, Mendel’s discoveries
transformed conventional breeding into an applied science positively correlated with the improvements in agricultural performance and quality (Hossfeld et al., 2017). While unequivocally successful, traditional plant breeding has its limitations. For example, it is constrained by fertilization incompatibility between plant species, the long generation time of crops, the extensive backcrossing required and the lack of efficient methods of selection. Moreover, when plants are crossed, undesirable traits can also be transferred along with the trait of interest (Breseghello and Coelho, 2013).
Mutations have also been exploited for crop improvement. These mutations can naturally occur spontaneously at a very slow rate, but they can also be induced by chemicals or by physical methods. For instance, mutagenesis methods such as irradiation (x-rays and gamma-rays) of seed and plant germplasm have been exploited to generate random mutations to obtain desirable phenotypes for crop improvement. Interestingly, plant varieties generated by random mutagenesis are exempt from regulatory restrictions, licensing costs and societal prejudice framing genetically modified organisms (GMOs) since the induced mutations could occur naturally. However, mutation breeding also has its limitations. It causes highly unpredictable DNA damage and selection of an agronomically relevant crop mutant is an extremely laborious and time-consuming process (Georges and Ray, 2017).
As the world’s population continues to rise, a new revolution in agriculture is urgently needed to reverse the declining trend in global food security. New targeted mutagenesis technologies could offer more reliable means to accelerate the pace of agricultural research and innovation
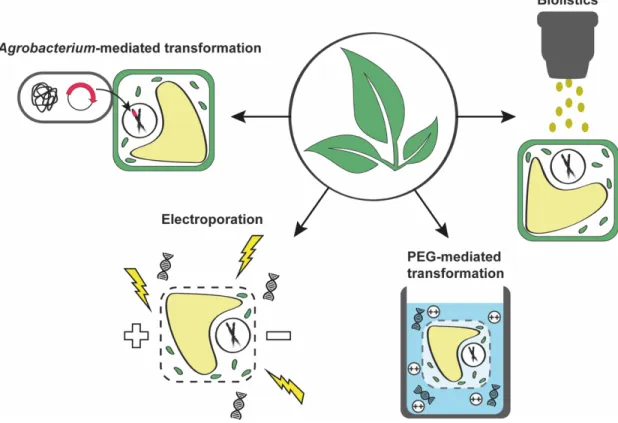
(Liu et al., 2013). The groundbreaking discovery of site-directed nucleases, or molecular scissors, and the ability to deliver foreign genes into the nucleus of plant cells led to the development of powerful gene modification techniques. Meganucleases, zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) have all proven useful for site-directed gene modification. More recently, clustered regularly interspaced short palindromic repeats (CRISPR, pronounced “crisper”) and CRISPR associated (Cas) genes coding for proteins working together with CRISPR were recognized as a powerful asset to the genetic toolbox due to their increased efficiency, versatility and accessibility (Wright et al., 2016). Modern sequencing and genome assembly technologies as well as big data analyses also enable rational crop improvement and testing. The characterization of genetic variations and their association with agronomically relevant traits is now feasible at an unprecedented level (Bevan et al., 2017). The CRISPR-Cas9 (with CRISPR-associated protein 9) gene editing tool offers an important alternative for overcoming the limitations of traditional breeding techniques. It has proven useful for rapidly creating desirable phenotypes in plants, but its adoption has been controversial.
According to the U.S. Department of Agriculture (USDA), plant varieties developed using precise gene-editing technologies like CRISPR-Cas9 can be commercially grown without being regulated as GMO (USDA, 2018), whereas these crops are subjected to GMO laws in the European Union (Callaway, 2018). A negative emotional response is associated with the GMO label although scientific evidences demonstrate the safety and positive impact of GMOs. For example, they have reduced chemical pesticide use, boosted crop yields and increased farmer profits significantly, including in developing countries (Klumper and Qaim, 2014). The opposition to GMO is often driven by fear and misinformation. This chapter explains how genes are modified, and we hope the readers will gain knowledge to form their own opinions and speak out on the issue in an informed way. Of note, in the next paragraphs, we will explain the concepts behind the CRISPR-Cas9 technology. For readers not highly familiar with this branch of biology, we encourage you to read the text while simultaneously glancing at the figures and the abbreviation list.
The microbial origin of CRISPR-Cas9
About half of the known bacteria possess a segment of genomic material consisting of a CRISPR region and cas genes (Burstein et al., 2016; Haft et al., 2005; Jansen et al., 2002; Mojica et al., 2005). In these microbes, CRISPR-Cas act as a natural defense mechanism against invading nucleic acids, such as DNA or RNA from bacteriophages (bacterial viruses called "phages" for short) and plasmids (mobile genetic elements). These systems are highly diverse. To-date, there are 2 classes, 6 types and various subtypes of CRISPR-Cas systems classified according to their associated Cas genes/proteins (Makarova et al., 2015; Shmakov et al., 2017). Recent analyses of CRISPR-Cas systems in uncultivated microbes suggest that the currently known systems are just the tip of the iceberg and a vast number likely remain to be discovered (Burstein et al., 2017). Class 1 systems employ a multi-subunit cleavage complex composed of numerous Cas proteins whereas class 2 systems are much simpler and employ a single multidomain Cas protein. One of the class 2 systems, type II CRISPR-Cas, is characterized by the presence of the signature protein Cas9, the widely used genome editing enzyme (Koonin et al., 2017).
The first experimental demonstration of the adaptive immunity provided by CRISPR-Cas systems in bacteria was reported just over a decade ago (Barrangou et al., 2007).
Streptococcus thermophilus, a bacterium used by the dairy industry to make cheese and
yogurt, was shown to naturally respond to phage infection by incorporating a short DNA fragment (named a spacer) from the invading phage genome into its CRISPR region. Transcription and maturation of the CRISPR region lead to the production of several small CRISPR RNAs (crRNAs) that, individually, form a duplex with a trans-activating CRISPR RNA (tracrRNA) (Deltcheva et al., 2011). The RNA duplex becomes a tracking device as any given crRNA can bind a sequence-matching DNA (Brouns et al., 2008). Because the RNA duplex also binds to the nuclease Cas9, the tracking device can bring the molecular scissor to its matching DNA target. As such, this “surveillance/interference complex” (crRNAs + Cas9) arms the microbes to defend against subsequent invasion by foreign DNA containing a region matching the crRNA.
Interestingly, Cas9 was reported to be the only protein required for the interference process (on-target DNA cleavage) of the S. thermophilus type II CRISPR-Cas system (Garneau et
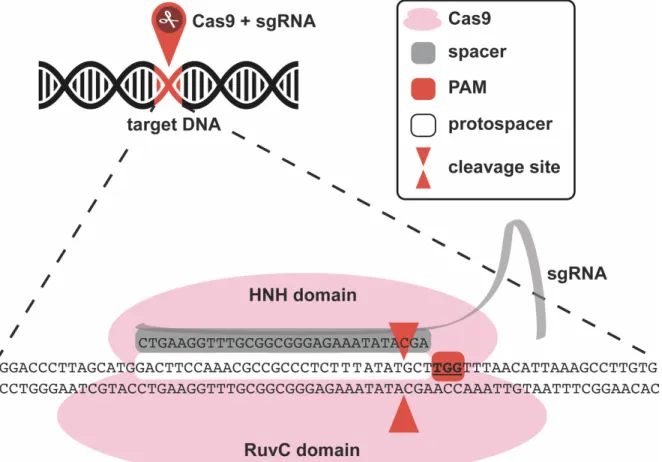
al., 2010; Sapranauskas et al., 2011). It is the spacer in the crRNAs that dictates the specificity of the system by annealing to the corresponding sequence (protospacer) on a DNA target. Moreover, a very specific and short motif (PAM; protospacer adjacent motif) is required next to the protospacer for Cas9 to cut (Bolotin et al., 2005; Deveau et al., 2008; Mojica et al., 2009). If these requirements are met, both strands of the DNA target will be cleaved by the two catalytic domains of Cas9; HNH and RuvC. The HNH domain, named for characteristic histidine (H) and asparagine (N) residues, cleaves the strand complementary to the spacer and the RuvC domain, named after a bacterial protein involved in DNA repair, cleaves the non-complementary strand (Nishimasu et al., 2014) (Figure 4).
The acquisition of a spacer in the CRISPR region, called adaptation, is akin to immunization, although hereditary in nature. Typically, during cell division, each daughter cell inherits an identical copy of the parent cell’s DNA, including the CRISPR region. This heredity means that the daughter cells can defend themselves against viruses they have not yet encountered (Fineran and Charpentier, 2012). Today, biotech industries leverage CRISPR immunization by exposing relevant bacterial strains to specific phages, making them more robust. This “vaccination” can even be improved by exposure to defective phages (Hynes et al., 2014). Across the globe, people have been consuming CRISPR-enhanced fermented dairy products for years as this natural system protect the “good” dairy bacteria from their natural predators, namely phages. This microbial system can now be harnessed to ensure sustainable and climate-compatible food systems.
Programmable DNA cleavage
Guided by a crRNA-tracrRNA duplex, Cas9 proteins naturally scan DNA for a specific PAM and bind their target protospacer through complementary base pairing with the spacer. Interestingly, Cas9 can be customized to target any DNA sequence upstream of a PAM by modifying the 20-nucleotide spacer sequence in the crRNA (Gasiunas et al., 2012). To make the system even simpler, the crRNA and the tracrRNA have been fused together to generate a single guide RNA (sgRNA) (Jinek et al., 2012) (Figure 4). Additionally, several sgRNAs with different spacers can be co-expressed to target multiple genes simultaneously (Cong et al., 2013). The possibility of multiplexing the CRISPR-Cas9 system is an important advantage over meganucleases, ZFNs and TALENs. For example, only a single
CRISPR-Cas9 construct was needed to mutate 14 different genes at once in the model plant
Arabidopsis thaliana (Peterson et al., 2016).
Figure 4. Cas9 site-directed nuclease
Cas9 is the molecular scissor programmed to cleave a precise genomic region when paired with a sgRNA. The interchangeable spacer in the sgRNA dictates the specificity of the system by annealing to the corresponding sequence (protospacer) on the target DNA if that sequence is adjacent to a PAM. The catalytic domains of Cas9, HNH and RuvC cleave both DNA strands, 3 nucleotides upstream of the PAM.
Only a few years after CRISPR-Cas9 was reported to function in several bacterial species (Sapranauskas et al., 2011), the system was successfully repurposed to edit the genome of eukaryotic cells (Cong et al., 2013; Mali et al., 2013). Currently, Cas9 from Streptococcus
pyogenes (SpCas9) is the most intensely studied Cas protein and the most widely used for
gene modification in many different plant species (Demirci et al., 2018). A key feature of SpCas9 is that it requires a 5’-NRG (where N = A, C, G or T and R = A or G) PAM to target