T
T
H
H
È
È
S
S
E
E
En vue de l'obtention du
D
DO
OC
C
TO
T
OR
RA
AT
T
D
DE
E
L
L’
’U
UN
NI
IV
VE
E
RS
R
SI
IT
TÉ
É
D
DE
E
T
T
OU
O
UL
L
OU
O
US
SE
E
Délivré par l'Université Toulouse III - Paul SabatierDiscipline ou spécialité : Physiopathologie expérimentale
JURY
Mr François Alhenc-Gelas (DR1 INSERM UMRS 872, Paris), Rapporteur Mr Christos Chatziantoniou (DR1 INSERM U702, Paris), Rapporteur Mr Jean-Jacques Helwig (DR1 INSERM UMRS 727, Strasbourg) Examinateur
Mme Hélène Hannaire (PU-PH, Toulouse), Examinateur
Mr Jean-Pierre Girolami (DR1 INSERM U858, Toulouse), Co-directeur de thèse Mr Ivan Tack (PU-PH, Toulouse), Co-directeur de thèse
Ecole doctorale : Biologie - Santé - Biotechnologies
Unité de recherche : Laboratoire de Physiologie et INSERM U858 Directeur(s) de Thèse : Dr Jean-Pierre Girolami et Pr Ivan Tack Rapporteurs : Mr François Alhenc-Gelas et Mr Christos Chatziantoniou
Présentée et soutenue par Marie Buléon Le 24 avril 2008
Titre : Physiologie rénale du récepteur B2 de la bradykinine : de la
Ecole Doctorale Biologie - Santé - Biotechnologie
T H E S E
Présentée et soutenue en vue de l’obtention du grade de
DOCTEUR DE L’UNIVERSITE TOULOUSE III
Discipline : Physiopathologie expérimentale
par
Marie BULEON
Physiopathologie rénale du récepteur B2 de la bradykinine : de
la néphropathie diabétique au choc septique
Directeurs de thèse : Dr Jean-Pierre Girolami et Pr Ivan Tack
Soutenue le : 24 avril 2008
Devant le jury composé de :
Mr François Alhenc-Gelas, DR1 INSERM UMRS 872, Paris Rapporteur
Mr Christos Chatziantoniou DR1 INSERM U702, Paris Rapporteur
Mr Jean-Jacques Helwig DR1 INSERM UMRS 727, Strasbourg Examinateur
Mme Hélène Hannaire Professeur, Toulouse Examinateur
Mr Jean-Pierre Girolami DR1 INSERM U858, Toulouse Co-directeur de thèse
Tout d’abord, je tiens à remercier les membres du jury, les Dr François
Alhenc-Gelas, Christos Chatziantoniou et Jean-Jacques Helwig, pour l’intérêt qu’ils ont porté à
mon travail, le temps qu’ils ont consacré à l’examiner et pour les remarques et suggestions qu’ils m’ont apportées.
Je souhaite remercier avec beaucoup de reconnaissance mes 2 directeurs de thèse pour m’avoir permis de réaliser ce travail.
Pr Ivan Tack qui m’a accueilli dans son équipe et m’a accordé sa confiance il y a
déjà plus de 5 ans et qui m’a permis d’arriver jusque là. Merci pour toutes les connaissances que tu m’as apportées, pour ton soutien et ta confiance.
Dr Jean-Pierre Girolami qui m’a soutenue depuis le DEA, pour tout ce qu ‘il m’a
appris sur les kinines. Merci de ton aide et de ta disponibilité au cours de ces années et particulier pour la rédaction de la thèse et la préparation de la soutenance.
Ce travail a été réalisé principalement dans le Laboratoire de Physiologie de la Faculté de Médecine, je remercie le Pr Jean-Louis Ader d’avoir accepter de m’y accueillir.
Je remercie les membres de l’équipe sans qui ce travail n’aurait pas été possible et qui ont partagé mon quotidien au labo pendant ces 5 années
Françoise Praddaude pour tout ce qu’elle m’a appris, pour son aide et sa gentillesse.
Merci de ton soutien qui m’a été précieux tout le long de mon séjour au labo et surtout à certains moments difficiles. J’ai apprécié les longues discussions autour des repas de midi partagés au labo.
Julien Allard qui est arrivé dans le labo en même temps que moi, avec qui j’ai
partagé plusieurs années de manip. Merci pour ton aide, tes conseils et tes encouragements, j’ai beaucoup apprécié de travailler avec toi.
Aux autres personnes qui font le quotidien du Laboratoire de Physiologie, Maïtée
Ranera, Mohamed Khetta, Marie-Pierre Groussous, Marie-José Fouque, Marie-Claire Mbongo, vous avez tous participé d’une façon ou d’une autre à mon travail. Merci de votre
aide, de votre bonne humeur et pour tous les bons moments passés ensemble.
Acil Jaafar pour le travail sur l’Irbésartan que j’ai apprécié de partagé avec toi. Bon
courage pour ta thèse et la suite.
Thierry Seguin pour tout le travail sur le choc septique.
Même si on se voit moins souvent, je n’oublie pas Christiane Pecher pour sa disponibilité, son aide et les dépannages en tout genre pour les manip ; et Nelly Blaes arrivée récemment dans l’équipe, merci pour ton soutien et tes conseils pendant les dernières étapes de la thèse.
A toutes les personnes de l’ INSERM pour leur sympathie et les coups de mains qu’ils m’ont donné..
Enfin, merci à tous ceux qui partagent ma vie.
Ma famille, qui m’entoure et me soutient (merci à mes parents de m’avoir permis de faire de si longues études).
Mes amis pour tous les bons moments qui permettent de décompresser et se changer les idées.
A Matthieu, simplement pour être là, merci pour ton soutien et ton écoute, ces années ont été beaucoup plus faciles en étant à tes côtés.
Et bien sûr à Maël, qui a pointé son petit nez pendant ma thèse, pour l’immense bonheur qu’il m’apporte.
Table des matières
INTRODUCTION... 10
PARTIE I : BRADYKININE ET REIN... 14
I. ORGANISATION DU SYSTEME KALLICREINE-KININE... 15
I.1. Composants de la cascade paracrine ... 15
I.1.a. Composants du système kallicréine-kinine... 15
I.1.b. Interactions entre le système rénine-angiotensine et le système kallicréine-kinine ... 16
I.2. Des récepteurs des kinines aux actions physiologiques... 18
I.2.a. Récepteurs B1 et B2 ... 18
I.2.b. Localisation et régulation des récepteurs B1 et B2... 19
I.2.c. Signalisation et effets... 20
II. UNE VISION FONCTIONNELLE ET INTEGREE DU SYSTEME KALLICREINE-KININE... 22
II.1. Régulation de la pression artérielle ... 23
II.2. Inflammation et douleur... 24
II.3. Physiopathologie cardiovasculaire... 25
II.4. Effets sur la prolifération et la fibrose... 25
III. SYSTEME KALLICREINE-KININE : DE LA PHYSIOLOGIE A LA PATHOLOGIE RENALE... 27
III.1. Localisation et actions physiologiques de la bradykinine... 27
III.1.a. Localisation rénale des composants du système kallicréine-kinine... 27
III.1.b. Actions rénales de la bradykinine ... 28
III.2. Modifications expérimentales du système kallicréine-kinine ... 29
III.2.a. Transfert du gène de la kallicréine ... 29
III.2.b. Invalidation du gène de la kallicréine ... 31
III.2.c. Surexpression/invalidation génétique du récepteur B2 ... 31
III.2.d. Invalidation du récepteur B1... 33
III.2.e. Invalidation des récepteurs B1 et B2... 33
III.2.f. Inhibition pharmacologique du système kallicréine-kinine ... 34
III.3. Contribution personnelle : Profil fonctionnel rénal chez les souris KOB2, évidence d’un rôle tonique du Récepteur B1 surexprimé ... 35
III.4. Place de la bradykinine dans les effets du blocage du système rénine-angiotensine... 38
III.4.a. Inhibiteurs de l’Enzyme de Conversion ... 38
III.4.b. Antagonistes du récepteur AT1 de l’angiotensine II (ARA2) ... 41
IV. CONCLUSION... 43
PARTIE II – NEPHROPATHIE DIABETIQUE ET SYSTEME KALLICREINE-KININE ... 44
I. UN CHALLENGE SANITAIRE... 44
II. DE LA PHYSIOPATHOLOGIE A LA THERAPEUTIQUE... 45
II.1. Les médiateurs à l’échelon cellulaire ... 46
II.1.a. Rôle de l’hyperglycémie / AGE ... 46
II.1.b. Rôle du stress oxydant ... 48
II.1.c. Facteurs de croissance ... 48
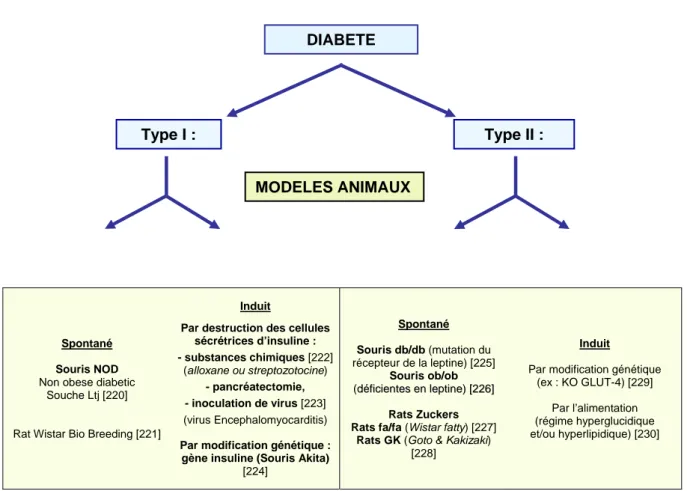
II.2. Intérêt et apport des modèles animaux ... 52
II.3. Thérapeutiques actuelles et perspectives ... 56
II.3.a. Définir une stratégie ... 56
II.3.b. La fibrose rénale est-elle réversible ?... 57
II.3.c. Les approches pharmacologiques ... 58
II.3.d. Autres cibles thérapeutiques potentielles ... 60
II.3.e. De l’approche protéomique à la thérapie génique / cellulaire... 61
III. TRAITEMENTS ET PROTECTION RENALE : DU BLOCAGE DU SRA A LA MISE EN JEU DU SKK... 63
III.1. Blocage du système rénine-angiotensine ... 63
III.2. Effets collatéraux du blocage du SRA : mise en jeu du SKK et autres... 63
III.3. Activation du récepteur B2 : une nouvelle perspective de néphroprotection... 65
III.3.a. Arguments pour le rôle protecteur de la bradykinine ... 65
III.3.b. Mécanismes potentiels de l’effet néphroprotecteur... 67
III.4. Quelle place pour le récepteur B1 ? ... 71
IV. CONTRIBUTION PERSONNELLE : PLACE DU RECEPTEUR B2 DANS LES EFFETS DU BLOCAGE DU SRA AU COURS DE LA NEPHROPATHIE DIABETIQUE EXPERIMENTALE... 72
PARTIE III – CHOC ENDOTOXINIQUE ET SYSTEME KALLICREINE-KININE... 84
I. COMPRENDRE POUR AMELIORER LE PRONOSTIC... 84
I.2. Mécanismes généraux... 85
I.3. Options thérapeutiques actuelles ... 88
II. QUELLE EST LA PLACE DU SYSTEME KALLICREINE-KININE ? ... 90
II.1. Rôle du recepteur B1 ... 91
II.2. Rôle du recepteur B2 ... 93
II.3. Différences entre les actions des deux récepteurs ... 94
III. SYSTEME KALLICREINE-KININE ET HEMODYNAMIQUE RENALE AU COURS DU CHOC ENDOTOXINIQUE. ... 95
IV. CONTRIBUTION PERSONELLE : IMPLICATION DES RECEPTEURS B1 ET B2 DANS L’HEMODYNAMIQUE SYSTEMIQUE ET RENALE AU COURS DU CHOC ENDOTOXINIQUE EXPERIMENTAL... 97
V. SYSTEME KALLICREINE-KININE ET CHOC SEPTIQUE, NOUVEAU MODELE, NOUVEAUX OUTILS ET NOUVELLES PERSPECTIVES... 98
CONCLUSIONS - PERSPECTIVES ... 99
Table des figures et tableaux
FIGURE 1 : ORGANISATION DES SYSTEMES RENINE-ANGIOTENSINE ET KALLICREINE-KININE... 17
FIGURE 2 : EFFETS SUR LES PARAMETRES GENERAUX... 36
FIGURE 3 : PARAMETRES FONCTIONNELS RENAUX... 37
FIGURE 4 : POSSIBILITES D’ACTIVATION DU RB2 DURANT UN TRAITEMENT PAR UN IEC OU UN ARA2 ... 41
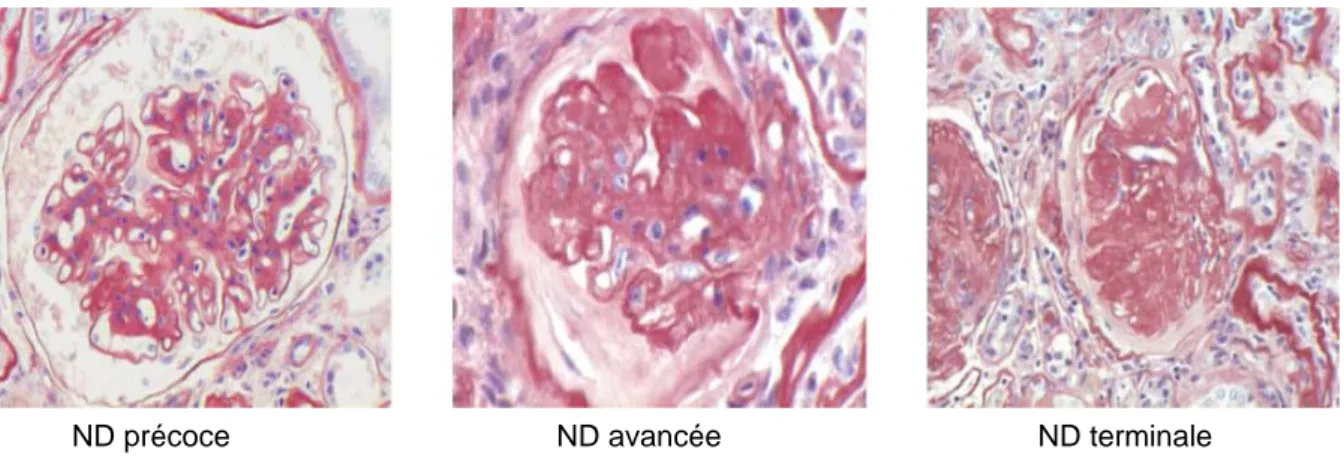
FIGURE 5 : EVOLUTION DE LA GLOMERULOSCLEROSE. GLOMERULES HUMAINS EN MICROSCOPIE OPTIQUE A DIFFERENTS STADES DE NEPHROPATHIE DIABETIQUE ... 45
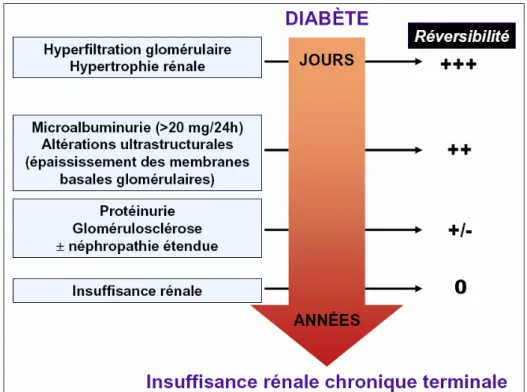
FIGURE 6 : EVOLUTION DE LA NEPHROPATHIE DIABETIQUE CHEZ L’HOMME... 46
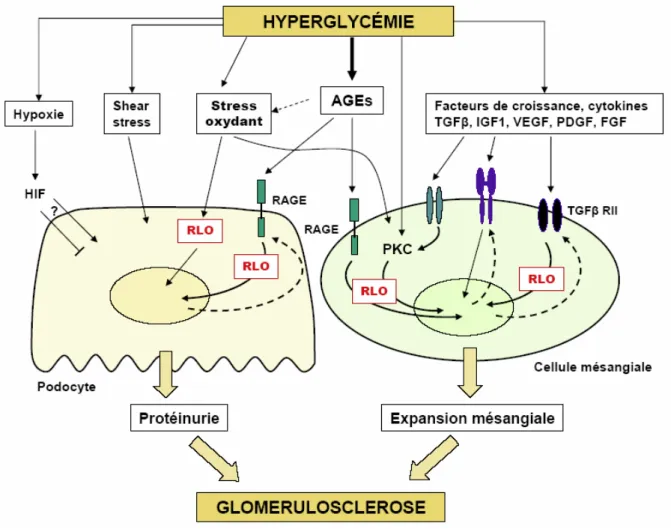
FIGURE 7 : PRINCIPAUX MECANISMES PHYSIOPATHOLOGIQUES DE LA NEPHROPATHIE DIABETIQUE ... 51
FIGURE 8 : EXEMPLES DE MODELES EXPERIMENTAUX DE DIABETE CHEZ LE RAT ET LA SOURIS... 52
FIGURE 9 : DIFFERENTS MECANISMES POSSIBLES DE NEPHROPROTECTION ASSOCIES A L’ACTIVATION DU RECEPTEUR DE LA BRADYKININE... 70
FIGURE 10 : DEFAILLANCE RENALE AU COURS DU SEPSIS... 88
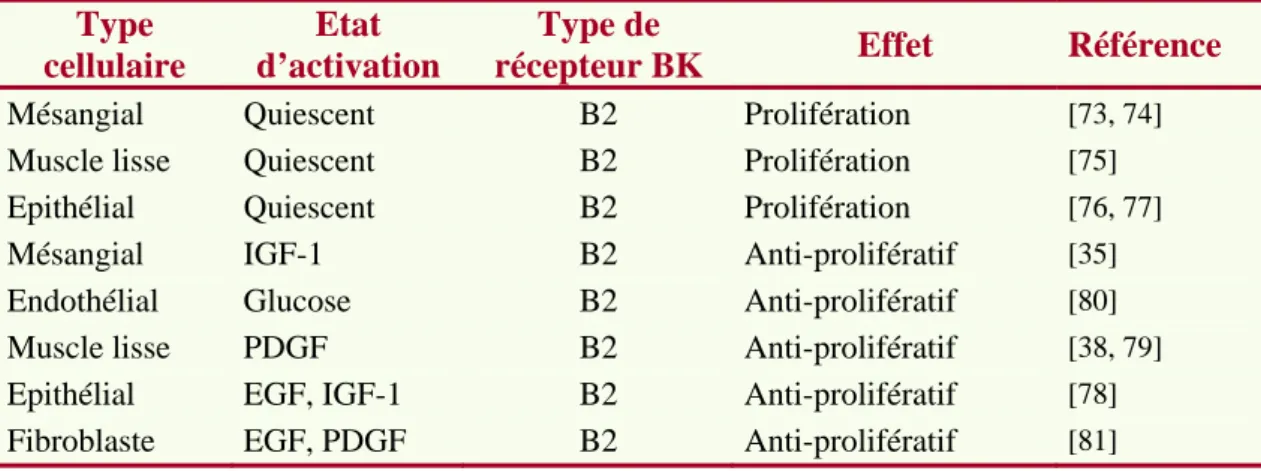
TABLEAU 1: PRINCIPAUX TRAVAUX IN VITRO SUR L'EFFET PROLIFERATIF OU ANTIPROLIFERATIF DE LA BK ... 26
TABLEAU 2 : APPORT DES QUELQUES MODELES GENETIQUEMENT MODIFIES DANS LA CONNAISSANCE DE LA GLOMERULOSCLEROSE DIABETIQUE... 55
Abréviations utilisées
AGE Advanced Glycation End-Product
AMPc Adénosine Mono-Phosphate cyclique
APC Activated Protein C
ARA2 Antagonistes des Récepteurs AT1 de l’Angiotensine II
AT1 Récepteur de type I de l’Angiotensine II
AT2 Récepteur de type II de l’Angiotensine II
BK Bradykinine
BMP7 Bone Morphogenic Protein 7
CTGF Connective Tissue Growth Factor
DAG Diacylglycérol
DFG Débit de Filtration Glomérulaire
DID Diabète Insulino-Dépendant
DNID Diabète Non Insulino-Dépendant
ECA Enzyme de Conversion de l’Angiotensine
ECA 2 Enzyme de Conversion 2 de l’Angiotensine 2
EGF Epithelial Growth Factor
ENaC Epithelial Na2+ Channel
FGF Fibroblast Growth Factor
GMPc Guanosine Mono-Phosphate cyclique
HGF Hepatocyte Growth Factor
HDAC Histone Desacethylases
HIF Hypoxia Inducible Factor
HOPE Heart Outcomes Prevention Evaluation
IEC Inhibiteurs de l’Enzyme de Conversion
IGF1 Insulin Growth Factor 1
IL Interleukine
IP3 Inositol 1,4,5-triphosphate
JNK Jun-N-terminal Kinase
KOB1 Souris invalidées pour le récepteur B1 de la bradykinine
KOB2 Souris invalidées pour le récepteur B2 de la bradykinine
L-NAME N(G)-nitro-l-Arginine Methyl Ester, inhibiteur de la NOS
LPS Lipopolysaccharide MAPK Mitogen Activated Protein Kinases
NFκB Nuclear Factor κB
NOD Non Obese Diabetic
NO Monoxyde d’azote
NOSe NO-Synthase endothéliale
NOSi NO-Synthase inductible
NOSn NO-Synthase neuronale
p27kip1 Inhibiteur des Cyclin Dependant Kinases
PAF Platelet-Activating Factor
PAI-1 Plasminogen Activator Inhibitor-1
PDGF Platelet-Derivated Growth factor
PGE2 Prostaglandine E2
PIP2 Phosphatidylinositol 4,5-biphosphate
PKC Protéine Kinase C
PLA2 Phospholipase A2
PLC Phospholipase C
RAGE Receptor for Advanced Glycation End-Product
RB1 Récepteur B1 de la bradykinine
RB2 Récepteur B2 de la bradykinine
RCPG Récepteur Couplé aux Protéines G
REIN The Ramipril Efficacy In Nephropathy
RLO Radicaux Libres Oxygénés
SDKP Peptide N-acetyl-seryl-aspartyl-lysyl-proline
SHR-SP Stroke-prone spontaneously hypertensive rats
SKK Système Kallicréine-Kinine
SOD Super Oxyde Dismutase
SRA Système Rénine Angiotensine
STZ Streptozotocine TGFβ Transforming Growth Factor β
TGFβ RII Récepteur de type II du TGFβ
TNF Tumor Necrosis Factor
TZD Thiazolidinediones
INTRODUCTION
Le diabète, de type I comme de type II, se complique fréquemment d’une atteinte rénale (prévalence ≥ 15 %), caractérisée principalement par une atteinte glomérulaire avec une protéinurie qui évolue vers l’insuffisance rénale. En dépit des efforts de contrôle de la glycémie, 50 % des diabétiques de type I développent une néphropathie après 20 ans de maladie. Ce problème est encore plus critique au cours du diabète de type II puisque, même si la durée de la maladie est souvent inférieure, il est pratiquement impossible d’obtenir un équilibre glycémique optimal. L’espérance de vie croissante dans ce cas explique l’incidence croissante des néphropathies diabétiques (ND) au cours du diabète de type II. Aujourd’hui, seul le blocage du Système Rénine Angiotensine (SRA) par les Inhibiteurs de l’Enzyme de Conversion (IEC) ou par les antagonistes du récepteur AT1 de l’angiotensine II, a montré une efficacité clinique pour ralentir la progression de la néphropathie diabétique. L’effet protecteur des IEC a longtemps été attribué exclusivement au blocage de la synthèse d’angiotensine II. Toutefois, un nombre croissant d’arguments expérimentaux suggère que la mise en jeu du Système Kallicréine-Kinine (SKK) contribue aussi à l’action bénéfique des IEC. La bradykinine (principale molécule du SKK) est un nonapeptide qui agit sur deux récepteurs couplés aux protéines G : les récepteurs B1 et B2. Le rôle du récepteur B2 (RB2) a été le plus étudié et sa contribution aux mécanismes d’action des IEC semble la plus importante. Bien avant la mise en évidence de son action trophique au cours des néphropathies chroniques, la BK a été impliquée dans des dysfonctionnements aigus du rein,
comme le choc septique dont l’incidence est d’ailleurs augmentée par la présence d’un diabète. Le système kallicréine-kinine occupe une place notable dans la physiopathologie du choc endotoxinique. Cependant son impact global, délétère ou protecteur, ainsi que les actions respectives des récepteurs B1 et B2 sont encore mal déterminés.
Nos travaux avaient pour but de préciser le rôle du récepteur B2 de la bradykinine au cours de l’insuffisance rénale chronique (néphropathie diabétique) ou aiguë (choc endotoxinique). L’idée principale était de rechercher de nouvelles cibles thérapeutiques potentielles pour protéger le rein dans ces deux pathologies.
Initialement, nous avons étudié l’effet du blocage du RB2 sur l’expression et l’activation de plusieurs facteurs de croissance au cours d’un traitement par un IEC chez le rat diabétique (Streptozotocine) (Allard J, Buléon M, Cellier E, Renaud I, Pecher C, Praddaude F, Conti M, Tack I and JP Girolami.ACE inhibitor reduces growth factor receptor expression and signalling but also albuminuria through B2-kinin glomerular receptor activation in diabetic rats. Am J Physiol Renal
Physiol. 2007 ; 293(4) : F1083-92). Ensuite, nous avons développé dans le Laboratoire le modèle des souris obèses et diabétiques de type II, C57BlKs db/db, qui développent des lésions rénales plus progressives et plus proches de celles observées chez l’Homme que le rat. Comme la néphropathie diabétique est avant tout une glomérulopathie, la première étape a consisté en l’adaptation chez la souris d’une méthode d’isolement glomérulaire par tri magnétique sélectif. Ce travail a fait l’objet d’une communication internationale (Buleon M, Allard J, Praddaude F, Ader JL, Tack I: Glomerular expression and activation of IGF-I pathway in type I and type II diabetes in mice. (poster) ASN 37th Annual Meeting & Scientific Exposition, St Louis,
Missouri, october 2004) et le manuscrit, en préparation, sera soumis prochainement à la revue
European Journal of Physiology. Nous avons ensuite étudié le rôle du récepteur B2 de la bradykinine (RB2) dans l’effet des IEC au cours de la néphropathie diabétique chez la souris
Christiane Pecher, Jean-Pierre Girolami and Ivan Tack. Pharmacological blockade of B2-kinin receptor reduces renal protective effect of Angiotensin-Converting Enzyme Inhibition in db/db mice model. Am J
Physiol Renal Physiol. 2008). Enfin, nous avons étudié dans le même modèle, l’effet du blocage du RB2 au cours d’un traitement cette fois par un antagoniste du récepteur AT1 de l’angiotensine II (ARA2) (Acil Jaafar, manuscrit en cours de rédaction).
L’extrême sensibilité des animaux diabétiques à l’action des IEC et au blocage du RB2 nous a amené à nous interroger sur les actions rénales du RB2 en conditions physiologiques et au cours du diabète. L’invalidation du RB2 a pour principale conséquence une diminution du débit de filtration glomérulaire. Nous avons surtout été frappés par le fait que les souris invalidées pour le RB2 surexpriment sensiblement le RB1 (la réciproque pour le KOB1 étant aussi exacte). Les conséquences observées dans le modèle KOB2 pourraient aussi résulter de la surexpression du RB1. L’utilisation d’antagonistes du RB1 et du RB2 dans les deux modèles de KO est en cours et fera prochainement l’objet d’une publication.
Le choc endotoxinique est une circonstance dramatique mais malheureusement assez fréquente (entre autre chez le diabétique), au cours de laquelle l’altération des fonctions rénales détermine largement le pronostic global du patient. Nous avons étudié les conséquences fonctionnelles rénales d’un choc endotoxinique induit par le lipopolysaccharide (LPS) chez des souris sauvages, KOB1 ou KOB2. (Seguin T, Buleon M, Destrube M, Ranera MT, Couture R, Girolami JP, Tack I. Hemodynamic and renal involvement of B(1) and B(2) kinin receptors during the acute phase of endotoxin shock in mice. Int Immunopharmacol. 2008 ; 8(2) : 217-21). Des travaux complémentaires sont en cours dans un modèle expérimental de choc septique par ponction caecale. Ceux-ci concernent l’implication des récepteurs de la bradykinine dans l’augmentation de la perméabilité capillaire, qui contribue à altérer le pronostic du choc septique.
La synthèse des résultats de nos travaux montre que le blocage du récepteur B2 de la bradykinine détermine une perte sensible d’efficacité des IEC. Plus encore, cela nous a permis d’étayer avec des arguments de plus en plus directs le concept de l’effet néphroprotecteur de l’activation du RB2. Cette réflexion a fait l’objet d’une revue faisant le point sur l’état des connaissances actuelles concernant le rôle potentiellement néphroprotecteur du RB2
(Bradykinine et néphroprotection, Pourquoi ? Comment ? Perspectives. Marie Buléon, Marion
Merhenberger, Christiane Pécher, Françoise Praddaude, Réjean Couture, Ivan Tack, Jean-Pierre
Girolami. MEDECINE/SCIENCES 2007 ; 23 : 1141-7). Finalement, il reste à vérifier cette
hypothèse par l’étude des effets directs d’un agoniste stable de la bradykinine. Nous disposons d’un des premiers agonistes non peptidiques (donc résistant à la dégradation endogène) de la bradykinine utilisable in vivo. Nous avons commencé à tester l’effet de cet agoniste, en priorité dans le modèle de la néphropathie diabétique.
PARTIE I : BRADYKININE ET REIN
Les kinines sont des médiateurs paracrines appartenant à une famille de peptides endogènes constitués de 9 à 11 acides aminés. Ce sont les peptides actifs du système kallicréine-kinine (SKK), dont le plus connu est la bradykinine (BK). Le SKK est un système protéique impliqué dans de nombreuses fonctions physiologiques telles que la régulation locale et systémique de la pression artérielle, la volémie, la perméabilité vasculaire, les réponses inflammatoires, la médiation de la douleur et le phénomène de coagulation. Le système comprend des substrats (les kininogènes), des enzymes (les kallicréines) et des peptides vasoactifs (les kinines), qui agissent sur deux récepteurs à sept domaines transmembranaires (les récepteurs B1 et B2), associés à l’activation d’un réseau de signalisation complexe.
La reconnaissance de ce système remonte aux travaux d’Abelous et Bardier (1909) [1] qui ont observé que l’injection intraveineuse d’urine humaine chez le chien provoquait une importante chute de la pression artérielle. Plus tard, en 1930 le groupe de E. Werle isola cette substance hypotensive, à partir d’extraits de pancréas et la désigna du nom de « kallicréine » [2]. La bradykinine a, quant à elle, été découverte 20 ans plus tard par le groupe de Rocha e Silva.
L’organisation du système kallicréine-kinine est à la fois complexe et classique pour un système de communication paracrine relativement ubiquitaire : un propeptide, une enzyme activatrice, 2 récepteurs de type RCPG et un catabolisme enzymatique très particulier.
I.
ORGANISATION DU SYSTEME KALLICREINE-KININE
I.1. COMPOSANTS DE LA CASCADE PARACRINE
I.1.a. Composants du système kallicréine-kinine
Les kinines sont libérées lors de l’hydrolyse enzymatique des kininogènes (substrats hépatiques circulants) par des sérine-protéases : les kallicréines. Il existe au moins deux types d’enzymes kallicréines : la kallicréine plasmatique et la kallicréine tissulaire. Elles diffèrent par leurs propriétés physico-chimiques et fonctionnelles, leurs substrats et leurs localisations [3]. Les deux hydrolysent, avec des affinités différentes, les kininogènes de haut et de bas poids moléculaire. La kallicréine plasmatique (PM : 60 kDa) est codée par un seul gène, elle est synthétisée au niveau du foie sous la forme d’un précurseur inactif, la prékallicréine, qui est la forme circulante. Après activation, elle hydrolyse essentiellement le kininogène de haut poids moléculaire (glycoprotéine synthétisée au niveau du foie) pour former la bradykinine. Les kallicréines tissulaires, codées par plusieurs gènes localisés chez l’Homme sur le chromosome 19 (PM entre 24 et 48 kDa), sont synthétisées dans de nombreux tissus, principalement le pancréas, le rein, l’intestin et le cerveau. Leur substrat préférentiel est le kininogène de bas poids moléculaire, à partir duquel elles vont libérer selon les espèces la bradykinine (Rat et Souris) ou la lys-bradykinine (ou kallidine chez l’Homme).
Les kininogènes de haut et de bas poids moléculaire sont produits dans le foie, ils résultent de l’épissage alternatif d’un seul gène [4]. Ils présentent des variations de séquence en fonctions des espèces, il existe en conséquence des différences entre espèces dans la séquence peptidique des kinines. La principale kinine, la bradykinine, est un nonapeptide de séquence : arg–pro–pro–gly–phe–ser–pro–phe–arg. Chez l’Homme, il existe deux autres
principales kinines : la kallidine ou lys-bradykinine (Lys-BK) qui est un décapeptide, et un nonapeptide, la des-Arg10-kallidine. Chez le Rat, il existe uniquement la BK et la des-Arg9 -BK ainsi qu’une kinine spécifique du rat : la T-kinine (ou Ile-Ser--BK), libérée essentiellement par l’action de la trypsine [5]. Toutes ces kinines agissent sur deux récepteurs couplés aux protéines G, nommés B1 et B2.
I.1.b. Interactions entre le système rénine-angiotensine et le système
kallicréine-kinine
In vivo, la BK est rapidement inactivée par deux voies enzymatiques principales
impliquant les kininases I et II. Sa demi-vie plasmatique est estimée à moins de 30 secondes. La kininase I, également connue sous le nom de carboxypeptidase N (CPN) dans le plasma ou carboxypeptidase M (CPM) sur la membrane plasmique des cellules vasculaires, génère la des-Arg9-BK. Celle-ci, agoniste préférentiel du récepteur B1, est responsable d’effets biologiques bien distincts de ceux de la BK. La kininase II, plus connue sous le nom d’Enzyme de Conversion de l’Angiotensine II (ECA), est la cible des Inhibiteurs de l’Enzyme de Conversion (IEC). C’est une métalloprotéase à doigts de zinc capable, d’une part de générer l’angiotensine II à partir d’angiotensine I et, d’autre part, de dégrader la BK en fragments BK1-5 et BK1-7 inactifs. L’ECA présente une plus grande affinité pour la BK que
pour l’angiotensine I [6]. Ce double rôle de l’enzyme de conversion, synthèse de l’angiotensine II et dégradation de la bradykinine, constitue un lien étroit entre ces 2 puissants systèmes vasomoteurs (Systèmes Rénine-Angiotensine, et SKK). L’organisation et l’intersection de ces deux systèmes sont représentées sur la Figure 1.
La bradykinine peut également être dégradée en BK1-7 par l’endopeptidase neutre. Les
inhibiteurs de vasopeptidases inhibent à la fois l’endopeptidase neutre et l’enzyme de conversion.
La très courte durée de vie de la BK laisse supposer qu’elle aurait plutôt une action locale, de type autocrine ou paracrine. En effet, les concentrations de BK tissulaires sont plus élevées que les concentrations plasmatiques [7]. De plus, les différents éléments nécessaires à la synthèse de BK sont présents dans la paroi de certaines artères [8, 9].
Figure 1 : Organisation des systèmes rénine-angiotensine et kallicréine-kinine
IEC : inhibiteurs de l’enzyme de conversion, ARA2 : antagonistes du récepteur AT1 de l’angiotensine II et IVP : inhibiteurs des vasopeptidases
I.2. DES RECEPTEURS DES KININES AUX ACTIONS PHYSIOLOGIQUES
I.2.a. Récepteurs B1 et B2
Les kinines agissent sur 2 récepteurs à 7 domaines transmembranaires (appartenant à la famille des récepteurs couplés aux protéines G), les récepteurs B1 et B2. Le récepteur B2 (RB2) a été cloné en 1991 par le groupe de Jarnagin [10], et le récepteur B1 l’a été quelques années plus tard [11]. La structure de ces 2 récepteurs est typique des récepteurs couplés aux protéines G, avec 7 domaines transmembranaires, un domaine N-terminal extracellulaire et un domaine C-terminal intracellulaire. Chez l’Homme, les gènes de ces deux récepteurs, localisés sur le chromosome 14 sont séparés par seulement 12 kb [12]. L’homologie de structure entre les récepteurs B1 et B2 n’est que de 36% chez l’Homme, et de 30% chez la Souris.
Initialement ces deux récepteurs ont été distingués et classés selon leurs propriétés pharmacologiques, en particulier selon leur affinité pour 2 différents ligands : la des-Arg9 BK et la BK [13].
Le RB1 est activé préférentiellement par la des-Arg9-BK et la des-arg10-lys-BK, métabolites endogènes de la bradykinine et de la lys-bradykinine respectivement après action de la kininase I. Il peut également être activé par la BK avec une moindre efficacité. Le récepteur B2 (RB2), responsable de la majorité des effets physiologiques décrits de la BK [3], est quand à lui activé exclusivement par la BK et la lys-BK [14].
I.2.b. Localisation et régulation des récepteurs B1 et B2
Le récepteur B2 (RB2) est constitutivement exprimé à la surface de nombreux types cellulaires, notamment les cellules endothéliales et musculaires lisses [15], les fibroblastes, les cellules mésangiales et épithéliales. Après liaison de la BK avec le récepteur et activation de la protéine G, le ligand est dissocié et le complexe récepteur/protéine G est rapidement internalisé. Ce processus implique la phosphorylation de plusieurs résidus sérine et thréonine dans le domaine C-terminal du récepteur [16]. Ce phénomène de désensibilisation permet d’éviter la suractivation des récepteurs B2 qui pourrait avoir des conséquences délétères au cours de la réponse inflammatoire, de la douleur ou bien pour le contrôle de la pression sanguine artérielle. De plus ce mécanisme d’endocytose permet secondairement le recyclage des RB2 à la membrane.
Contrairement au RB2, le RB1 est un récepteur essentiellement inductible. De ce fait, il est peu détectable dans les conditions physiologiques mais fortement exprimé au cours de certaines situations pathologiques [17]. Son expression est induite en cas de lésions tissulaires, en présence d’endotoxines bactériennes [18], et au cours de certaines pathologies comme le diabète. Cette induction se fait au niveau transcriptionnel et implique des cytokines inflammatoires telles que l’interleukine-1β et le TNFα [19, 20], ou encore l’activation des MAP-kinases, la mise en jeu du facteur de transcription nucléaire NFκB et la protéine kinase p38 [21-23].
A la différence du RB2, le RB1 est associé à une lente dissociation du ligand, il n’est ni désensibilisé ni internalisé [24, 25].
I.2.c. Signalisation et effets
Les récepteurs B1 et B2 sont associés à de multiples systèmes de signalisation complexes.
Ainsi, le RB2 active les voies de signalisation classiquement associées aux RCPG, impliquant le recrutement des protéines Gαq et Gαi, l’activation des phospholipases C et A2 et la formation des seconds messagers correspondants. Ces cascades de signalisation diffèrent suivant le type cellulaire concerné. La phospholipase C hydrolyse le phosphatidylinositol 4,5-biphosphate (PIP2) pour former les seconds messagers inositol 1,4,5-triphosphate (IP3) et diacylglycérol (DAG). L’IP3 provoque une augmentation rapide des concentrations intracellulaires en Ca2+, et est par exemple responsable de l’activation de la NO-synthase endothéliale et donc de la synthèse et de la libération de monoxyde d’azote (NO) [26]. Le NO ainsi produit diffuse jusqu’aux cellules cibles où il détermine la formation de guanosine monophosphate cyclase (GMPc). De nombreux travaux montrent que le NO et le GMPc sont les facteurs vasodilatateurs responsables des effets des kinines sur le tonus vasculaire. Tout d’abord, une accumulation de GMPc dépendante du NO a été montrée dans les cellules du muscle lisse vasculaire [27]. L’administration d’un antagoniste non peptidique du récepteur B2 (HOE-140 ou Icatibant) provoque une réduction des taux de GMPc dans le liquide interstitiel rénal [28]. L’inhibition de la NO-synthase par le L-NAME induit une augmentation de pression artérielle plus faible chez les souris KOB2 que chez les souris contrôles [29]. Enfin, les souris transgéniques surexprimant le RB2 présentent une élévation des concentrations rénales et urinaires d’adénosine monophosphate cyclase (AMPc) et de GMPc ainsi qu’une augmentation des taux urinaires de NO [30], alors que les souris n’exprimant plus le RB2 excrètent moins de nitrites et ont une concentration urinaire de
GMPc plus faible [31]. Tous ces arguments plaident en faveur d’un effet vasodilatateur de la bradykinine via la production de NO et de GMPc. L’activation du RB2 peut également stimuler la phospholipase A2 (PLA2) et induire la synthèse d’acide arachidonique, précurseur de la prostacycline [32]. Celle-ci stimule la production d’AMPc, un autre puissant vasodilatateur.
La BK est également capable d’activer des voies de signalisation moins classiques. En effet, une interaction directe a été démontrée entre le RB2 et des enzymes telles que la phospholipase Cγ, la NO-synthase neuronale (NOSn), la NO-synthase endothéliale (NOSe) [33]. Cette interaction entre le RB2 et la NOSe permet le recrutement de la protéine kinase Akt et permet ainsi le contrôle de la phosphorylation de la NOSe et de la production de NO. Le RB2 pourrait également interagir avec la tyrosine phosphatase SHP2 [34], cette interaction serait impliquée dans l’effet antiprolifératif de la BK. Cette complexité des voies de signalisation activées par la BK fournit une explication à ses effets parfois contradictoires au cours des phénomènes de prolifération cellulaire et de fibrose. En effet, selon les conditions, la BK pourrait avoir une action proliférative ou anti-proliférative (Tableau 1 p26). Dans la cellule mésangiale, par exemple, la BK serait capable de stimuler ou d’inhiber la phosphorylation des MAP-kinases ERK1 et 2 en fonction de l’état d’activation cellulaire en réponse à divers facteurs de croissance [35].
Le RB1 interagit aussi directement avec les protéines Gq et Gi [36] et active la plupart des voies de signalisation mises en jeu par le RB2, dont l’hydrolyse de PIP2, l’augmentation de Ca2+ intracellulaire, la libération d’acide arachidonique et l’activation de la NOSe. Le RB1 exerce un rôle important dans les processus d’inflammation. A l’instar du RB2, l’activation du RB1 peut entraîner des effets prolifératifs [37] ou anti-prolifératifs [38]. L’effet prolifératif impliquerait une activation des MAP-kinases. Inversement, Dixon et al [38] ont montré que
un effet anti-prolifératif du RB1 dans les cellules musculaires lisses. Il serait dû à l’activation prolongée des MAP-kinases qui activeraient secondairement l’inhibiteur de cycle cellulaire p27kip1.
Les deux récepteurs empruntent donc des voies de signalisation similaires pour induire des effets différents en fonction des tissus et de leur régulation à court terme (internalisation, désensibilisation). Puisque ces récepteurs activent de façon générale le même ensemble de voies de signalisation, ils ne peuvent être distingués sur la base de leurs effets biologiques. Par contre, leur blocage par des antagonistes pharmacologiques spécifiques permet de bien distinguer les effets B1 des effets B2.
II.
UNE VISION FONCTIONNELLE ET INTEGREE DU SYSTEME
KALLICREINE-KININE
Les rôles biologiques des kinines sont nombreux et comprennent : 1) la contraction ou relaxation des muscles lisses des tractus intestinal, urogénital et respiratoire ; 2) la régulation des transports ioniques épithéliaux, de l’hémodynamique systémique et de la pression artérielle [39] ; 3) un rôle de neurotransmetteur dans le système nerveux central et 4) un rôle modulateur de certaines fonctions cellulaires, telles que la prolifération (mitogénèse) et, plus récemment, des phénomènes de fibrose [40-43]. En pratique, leurs rôles prédominants semblent, dans l’état actuel des connaissances, la médiation de l’inflammation et de la douleur ainsi que le contrôle de la pression artérielle [44].
II.1. REGULATION DE LA PRESSION ARTERIELLE
La bradykinine, via la libération de NO et de GMPc, est un puissant vasodilatateur. Des travaux effectués chez l’Homme et dans plusieurs modèles expérimentaux, suggèrent qu’une diminution de la synthèse de BK favoriserait le développement de l’hypertension artérielle. Des études épidémiologiques [45, 46] ont montré une relation inverse entre les taux urinaire et rénal de kallicréine et la sévérité de l’hypertension. De même, les études expérimentales ont rapporté une réduction de l’excrétion urinaire de kallicréine chez des rats hypertendus [47]. Ces observations supportent l’idée d’une relation entre le SKK et la régulation de la pression artérielle. Cependant la BK n’apparaît pas comme un déterminant important de la pression artérielle en conditions physiologiques. Elle jouerait plutôt un rôle protecteur contre les facteurs qui tendent à la modifier, en particulier à l’augmenter. En effet les souris invalidées pour le RB2 ne semblent pas spontanément hypertendues [48, 49], bien que quelques travaux [29, 50-52] du groupe de Maddedu indiquent que les souris KOB2 développent une augmentation modérée de la pression artérielle. Cependant ces résultats n'ont pas été confirmés par les travaux d’autres groupes. Par contre, ces souris déficientes en RB2 présentent une hypersensibilité à certains facteurs tenseurs, en particulier un régime hypersodé [29, 53, 54]. Cela suggère que l’activation du RB2 participerait essentiellement à la régulation de la pression artérielle en condition de « stress ». La BK pourrait donc jouer un rôle protecteur en maintenant normale la pression artérielle en cas de surcharge sodée, certainement par son effet vasodilatateur NO-dépendant, mais peut être aussi via son effet natriurétique. Lorsque l’hypertension artérielle se développe tout de même, la BK pourrait également limiter ses complications : hypertrophie cardiaque, lésions rénales…
II.2. INFLAMMATION ET DOULEUR
Contrairement aux situations précédemment évoquées, une surproduction de bradykinine est observée au cours des états inflammatoires. Dans ce cas, le blocage de ses récepteurs permettrait de réduire l’intensité de la réaction inflammatoire et de la douleur [55-61]. Les kinines sont des puissants agents vasoactifs. En condition inflammatoire ou lors de lésions tissulaires, elles sont responsables d’une vasodilatation artériolaire mais aussi d’une extravasation plasmatique par augmentation de la perméabilité plasmatique [39]. Les deux récepteurs, B1 et B2, ont été impliqués dans le développement de réponses inflammatoires locales dans des modèles d’arthrites aiguës et chroniques chez le rat [62-65]. Le récepteur B1, ainsi que son ligand endogène, sont produits de novo sur les sites d’inflammation ou de lésions tissulaires. Ce récepteur a été impliqué dans de nombreuses pathologies inflammatoires chroniques dont certaines formes d’hyperalgésie chronique [66].
Les kinines sont également capables de stimuler directement les terminaisons nerveuses sensorielles et ainsi de générer la douleur et l’inflammation neurogène [66, 67]. De plus, les composantes du SKK sont exprimées au niveau du système nerveux central. Le RB2 jouerait, à ce niveau là, un rôle dans la modulation de la douleur [68, 69].
Les deux récepteurs des kinines jouent donc un rôle important dans le développement des réponses inflammatoires et de la douleur, avec toutefois des effets bien distincts : le RB2 interviendrait plutôt dans les phases aiguës de l’inflammation et de la douleur, tandis que le RB1 serait plutôt mis en jeu dans leurs phases chroniques.
II.3. PHYSIOPATHOLOGIE CARDIOVASCULAIRE
Les kinines exercent également un rôle important en physiopathologie cardiovasculaire. La bradykinine, via le RB2, aurait une action cardioprotectrice, indépendante de son effet vasodilatateur. En effet, les animaux surexprimant la kallicréine sont plus résistants à l’ischémie-reperfusion cardiaque [70]. Dans la même situation, les souris invalidées pour le RB2 développent des lésions myocardiques et des arythmies plus sévères [71]. L’effet anti-hypertrophique du RB2 a également été décrit au cours d’une étude clinique chez l’Homme [72].
II.4. EFFETS SUR LA PROLIFERATION ET LA FIBROSE
La littérature récente propose de « nouveaux effets » de la bradykinine, en particulier dans le contrôle de la prolifération cellulaire et la synthèse de protéines matricielles. Ces effets, parfois contradictoires, pourraient mettre en jeu l’activation de voies de signalisations différentes des voies classiques et n’impliquant pas le recrutement d’une protéine G.
Il a été rapporté à la fois des effets mitotiques et des effets anti-mitotiques de la bradykinine. En effet, la BK, via l’activation de la PKC a un effet inducteur de la prolifération de faible intensité sur de nombreux types cellulaires [73-77]. A l’inverse, d’autres travaux montrent une inhibition de la prolifération de cellules cancéreuses mammaires [78] et des cellules musculaires lisses [79]. Ces effets apparemment contradictoires suggèrent l’existence d’une double voie de signalisation. En fait, l’effet prolifératif ou anti-prolifératif de la BK semble dépendre de l’état d’activation préalable des cellules par les facteurs de croissance. Le Tableau 1 regroupe plusieurs travaux qui montrent que les effets prolifératifs sont observés
sur les cellules quiescentes alors que les effets anti-prolifératifs l’ont été sur des cellules en prolifération.
Tableau 1: Principaux travaux in vitro sur l'effet prolifératif ou antiprolifératif de la BK Type
cellulaire
Etat d’activation
Type de
récepteur BK Effet Référence
Mésangial Quiescent B2 Prolifération [73, 74]
Muscle lisse Quiescent B2 Prolifération [75]
Epithélial Quiescent B2 Prolifération [76, 77]
Mésangial IGF-1 B2 Anti-prolifératif [35]
Endothélial Glucose B2 Anti-prolifératif [80]
Muscle lisse PDGF B2 Anti-prolifératif [38, 79]
Epithélial EGF, IGF-1 B2 Anti-prolifératif [78]
Fibroblaste EGF, PDGF B2 Anti-prolifératif [81]
De la même façon, la bradykinine pourrait exercer, selon les types cellulaires, des effets fibrosants ou anti-fibrosants. En effet, in vitro, l’activation du RB1 stimule la synthèse de collagène par les fibroblastes pulmonaires [82]. A l’inverse, la BK réduit la synthèse de fibronectine et de collagène par les fibroblastes cardiaques [83]. In vivo, la BK aurait plutôt un rôle anti-fibrosant, en inhibant l’accumulation cardiaque de collagène après un infarctus chez le rat [84]. La surexpression de la kallicréine chez le rat, permet également de réduire la fibrose consécutive à une hypertrophie cardiaque [85], un effet dépendant du RB2. Enfin, après obstruction urétérale, les souris invalidées pour le RB2 présentent une fibrose rénale tubulo-interstitielle deux fois plus sévère [42]. De plus, l’effet anti-fibrosant des IEC sur les cellules mésangiales humaines semble être dépendant au moins en partie du RB2 [86]. Cet effet anti-fibrosant de la BK reposerait sur la mise en jeu de la stimulation des activateurs du plasminogène, ce qui provoquerait une activation des métalloprotéases responsables de la dégradation de la matrice extracellulaire [42].
III. SYSTEME KALLICREINE-KININE : DE LA PHYSIOLOGIE A
LA PATHOLOGIE RENALE
III.1. LOCALISATION ET ACTIONS PHYSIOLOGIQUES DE LA BRADYKININE
III.1.a. Localisation rénale des composants du système
kallicréine-kinine
Tous les éléments du système kallicréine-kinine sont exprimés au niveau du rein. Un premier travail a montré l’existence de sites de liaison pour 3H-BK le long du néphron [87]. Ensuite, l’utilisation de la technique de microdissection couplée à des tests fonctionnels (calcium, sécrétion de prostaglandines) a permis de décrire la localisation très précise des récepteurs B1 et B2 le long du néphron [88, 89]. Les récepteurs B1 et B2 ont été étudiés au niveau glomérulaire tant in vivo [73] que sur des cellules mésangiales en culture [90, 91].
L’ARN messager de la kallicréine a été détecté au niveau glomérulaire et au niveau du tubule contourné distal [92]. Le kininogène de bas poids moléculaire, substrat utilisé par la kallicréine rénale, a d’abord été localisé dans le rein par immunofluorescence au niveau du tubule contourné distal et du canal collecteur chez l’Homme et le Rat [93]. Depuis, chez l’Homme, l’expression de l’ARN messager du kininogène de bas poids moléculaire et du récepteur B2 ont été détectés dans les cellules juxtaglomerulaires, les cellules mésangiales, l’épithélium de la capsule de Bowman, les tubules proximaux et distaux, l’anse de Henlé, le canal collecteur et au niveau des cellules endothéliales des vaisseaux sanguins [94].
L’expression, au niveau rénal, de tous les composants du SKK suggère une production locale de BK et l’implication du système dans la physiologie rénale et les phénomènes d’homéostasie afférents.
III.1.b. Actions rénales de la bradykinine
Les premiers travaux sur les actions de la bradykinine au niveau rénal ont décrit ses effets vasodilatateurs, diurétiques et natriurétiques. En plus de ses actions vasodilatatrices déjà détaillées, la BK exerce, principalement via le RB2, d’autres effets indépendants, en particulier diurétiques et natriurétiques qui impliquent soit l’inhibition de la synthèse d’AMPc [95, 96] soit la synthèse de NO [96-98].
Ces études suggèrent que l’inactivation du SKK favorise l’hypertension artérielle et le développement de ses complications rénales. Le SKK est donc rapidement apparu comme un système de défense contre le SRA vasoconstricteur.
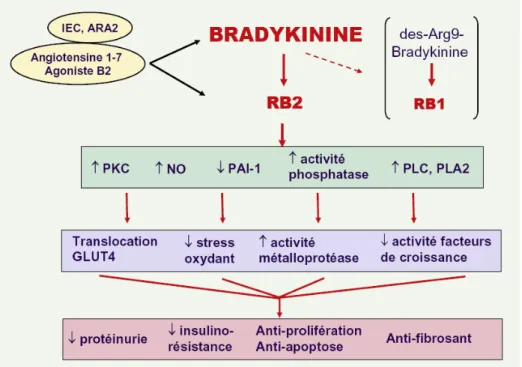
De nombreux arguments suggèrent que la BK exerce un rôle néphroprotecteur, indépendant de son effet sur l’hypertension. Ainsi, la perfusion de kallicréine chez le rat Dahl hypertendu, à une dose qui n’a pas d’effet sur la pression artérielle systémique, réduit la protéinurie, augmente le débit de filtration glomérulaire, et prévient le développement de la fibrose glomérulaire et tubulaire [99, 100]. Ces effets bénéfiques sont accompagnés d’une augmentation de l’excrétion urinaire de BK et de GMPc, ce qui confirme que l’effet de l’administration de kallicréine agit via l’augmentation de la synthèse de bradykinine [100]. De plus, ces effets étaient dépendants de l’activation de son récepteur B2 puisqu’ils étaient supprimés par la co-administration d’HOE-140 (antagoniste du RB2) [99]. Plus récemment, un travail utilisant le même modèle, a montré que l’administration directe de BK protège de l’atteinte rénale induite par un excès de sel [40]. Ce traitement a permis en effet de limiter la
protéinurie, la glomérulosclérose et la dilatation tubulaire, mais aussi de réduire l’accumulation de collagène, l’inflammation et l’apoptose dans le rein. Ces effets de la perfusion de BK étaient associés à une augmentation de la synthèse de NO, ainsi qu’à une diminution du stress oxydant, de l’expression du TGFβ et de l’activation des MAP-kinases.
Actuellement, de nombreuses études suggèrent un rôle néphroprotecteur de la BK. D’une part, elle occupe une place sensible dans l’effet protecteur rénal des IEC. D’autre part, sa capacité à réduire le développement de la fibrose suggère qu’elle pourrait avoir une action bénéfique directe au cours des pathologies rénales fibrosantes. Le développement de modèles de souris transgéniques pour les différents constituants du SKK, en particulier les souris invalidées pour le RB2, a permis d’apporter des preuves expérimentales très fortes en faveur du rôle néphroprotecteur de la BK dans différentes situations pathologiques.
III.2. MODIFICATIONS EXPERIMENTALES DU SYSTEME KALLICREINE-KININE
III.2.a. Transfert du gène de la kallicréine
La kallicréine, enzyme nécessaire à la synthèse des kinines, occupe une place essentielle dans le SKK. De façon intéressante, le débit d’excrétion urinaire de kallicréine urinaire chez l’homme est inversement corrélé à la sévérité des pathologies rénales [101]. Agir sur la disponibilité rénale de la kallicréine représente donc un moyen d’étudier les effets potentiellement néphroprotecteurs de la BK.
Le groupe de Chao a fait de nombreux travaux de thérapie génique expérimentale en transférant dans plusieurs modèles de néphropathie chez le rat, le gène de la kallicréine tissulaire humaine. Tout d’abord, dans un modèle de néphropathie induite par la gentamycine
[102], le transfert du gène de la kallicréine apportait un bénéfice fonctionnel (moindre diminution du DFG), et structural (ralentissement des lésions tubulaires et de la nécrose cellulaire épithéliale). Dans un modèle de néphrectomie 5/6 [103], ce même traitement réduisait la protéinurie et l’albuminurie et prévenait aussi le développement de lésions de sclérose glomérulaire et tubulaire. Cette méthode de thérapie génique par le gène de la kallicréine a montré les mêmes effets bénéfiques dans différents modèles de rats hypertendus développant des dommages cardio-vasculaires et rénaux [104-106]. Cet effet protecteur était accompagné d’une augmentation de la synthèse de NO et de GMPc, d’une diminution d’expression du TGFβ, de p27kip1, de la phosphorylation de la JNK et de ERK, mais aussi de la production d’ion superoxide. La kallicréine tissulaire protège donc du développement des complications rénales chez le rat hypertendu via une augmentation de la biodisponibilité du NO, la réduction du stress oxydant et de l’expression du TGFβ. Dans un travail récent, le même groupe a montré que le traitement par le gène de la kallicréine était capable non seulement de prévenir mais également de reverser la fibrose rénale en restaurant la production de NO et en inhibant celle d’ion superoxide [107, 108]. Il est intéressant de noter que, de façon inattendue et presque paradoxale, l’administration prolongée de kallicréine ne semble pas induire d’effets hypotenseurs.
III.2.b. Invalidation du gène de la kallicréine
L’invalidation du gène de la kallicréine glandulaire a été réalisée par le groupe de F. Alhenc-Gelas [109-111]. Chez ces animaux, la BK n’est plus détectable, ce qui indique que la kallicréine glandulaire est indispensable pour la synthèse de BK circulante. Ces souris sont normotendues et présentent une hémodynamique cardiaque et rénale normales [109]. Au niveau rénal, l’invalidation de la kallicréine tissulaire provoque une diminution de la réabsorption tubulaire du calcium [112], qui n’est pas retrouvée chez les souris invalidées pour le RB2, même en présence d’un antagoniste du RB1. Cela suggère un effet indépendant des kinines. Plus récemment, le même groupe a montré que les souris déficientes en kallicréine présentent un défaut d’activité du canal ENaC [113], suggérant un effet anti-natriurétique potentiel de la kallicréine via l’activation de ce canal sodé du néphron distal.
III.2.c. Surexpression/invalidation génétique du récepteur B2
Les souris transgéniques surexprimant le RB2 sont hypotendues [114], et présentent une augmentation du débit de filtration glomérulaire associée à une activation de la voie NO-GMPc/AMPc [30].
Par contraste, les souris déficientes en RB2 (KOB2), elles, ne sont pas hypertendues, et leur fonction rénale est normale [48]. D’autres travaux, contradictoires, ont montré que le KOB2 serait associé à une augmentation de la pression artérielle et de la fréquence cardiaque [29, 50-52]. Toutefois, cette observation n’a pas été confirmée par les travaux plus récents, il pourrait s’agir d’une simple variation des conditions expérimentales. Par contre l’absence de RB2 a un impact sensible dans certaines conditions pathologiques, ce qui étaye son rôle « protecteur » en terme d’équilibre homéostatique et d’hémodynamique rénale dans de telles situations pathologiques. Par exemple, les souris KOB2 présentent une hypersensibilité aux
régimes hypersodés [29, 53, 54]. En effet, un apport excessif de NaCl ainsi qu’un excès de minéralocorticoïdes [115] provoque une hypertension plus importante chez les souris KOB2 que chez des souris sauvages. Dans un autre cadre pathologique, les souris n’exprimant plus le RB2 développent une néphropathie diabétique plus sévère que la souche sauvage [116]. Cette observation est à rapprocher de l’observation selon laquelle les souris exprimant des copies multiples de l’enzyme de conversion (état associé à une forte dégradation de la BK et ainsi à une très faible activation du RB2) développent également une néphropathie diabétique plus sévère [117]. Enfin, la fibrose interstitielle après obstruction urétérale semble aggravée chez la souris n’exprimant plus le RB2 [42]. Ces résultats montrent que l’absence d’expression du RB2 ou sa faible activation aggrave le développement de lésions rénales. Cependant, l’utilisation de modèles de souris KO pour l’un des récepteurs de la BK présente une limite. En effet, plusieurs travaux ont montré que le récepteur non invalidé (RB1 chez les animaux KOB2 et inversement) est surexprimé [118, 119]. Cette surexpression pourrait correspondre à une adaptation physiologique destinée à compenser l’absence de l’autre récepteur. Duka et al [118] ont montré cette surexpression du RB1 chez les souris KOB2, le RB1 pouvant alors compenser la perte du RB2 en exerçant à sa place des effets vasodilatateurs.
III.2.d. Invalidation du récepteur B1
Les souris invalidées pour le RB1 sont normotendues [120]. L’effet de l’invalidation du RB1 a été étudié au niveau cardiaque [120, 121], dans les phénomènes de douleur [122, 123] et au cours du choc septique (cf Partie III) mais aucun effet sur les fonctions et la structure du rein n’a à ce jour été rapporté.
III.2.e. Invalidation des récepteurs B1 et B2
Le groupe de O. Smithies a développé un modèle de souris déficientes pour les 2 récepteurs des kinines, et a étudié leur susceptibilité à l’ischémie-reperfusion rénale [124]. Ils ont observé que les souris invalidées pour le RB2 (KOB2) présentaient des lésions consécutives à l’ischémie-reperfusion (lésions histologiques, apoptose, insuffisance rénale) plus importantes que les souris sauvages. De plus, les souris invalidées pour les 2 récepteurs (KOB1B2) développent des lésions encore plus importantes, ce qui suggère un rôle également protecteur du RB1. Le stress oxydant, ainsi que l’expression des ARNm du TGFβ, du CTGF et de l’endothéline 1, trois gènes impliqués dans le développement des lésions consécutives à l’ischémie-reperfusion, ont été recrutés de façon concomitante avec des amplitudes variables : WT < KOB2 < KOB1B2. Le développement de ce double KO a permis de montrer que les deux récepteurs des kinines peuvent exerçer un rôle néphroprotecteur dans le modèle d’ischémie-reperfusion [124]. Cependant, une autre équipe qui a également développé ce double KO ne confirme pas les effets aggravants de l’absence conjointe du RB1 et du RB2, du moins dans un autre modèle expérimental, celui du choc septique [125].
III.2.f. Inhibition pharmacologique du système kallicréine-kinine
Comme l’invalidation génétique du RB2, le blocage pharmacologique du SKK, par l’aprotinine qui inhibe la kallicréine, ou par un antagoniste du RB2, ne modifie pas la pression sanguine que ce soit chez l’Homme ou chez le Rat [126, 127]. Par contre, elle augmente les effets vasopresseurs de la deoxycorticostérone et de l’angiotensine II [128-130]. Chez le rat SHR-SP, spontanément hypertendu, le RB1 est fortement induit [131]. L’inhibition pharmacologique du RB1 provoque une augmentation de la pression sanguine artérielle et de l’excrétion urinaire d’albumine, parallèlement à une augmentation de l’expression du TGFβ et du collagène de type II. L’induction du RB1 au cours de l’hypertension dans ce modèle permettrait donc, via ses effets vasodilatateurs et anti-fibrosants, de protéger le rein des conséquences délétères de cette hypertension. Par contre, l’induction du RB1 lors du choc septique a un effet presseur qui pourrait limiter l’hypotension dans la phase aiguë [132, 133]. Ces observations doivent être prises en considération avant d’envisager le blocage pharmacologique systématique du RB1 comme perspective thérapeutique.
III.3. CONTRIBUTION PERSONNELLE : PROFIL FONCTIONNEL RENAL CHEZ LES SOURIS KOB2, EVIDENCE D’UN ROLE TONIQUE DU RECEPTEUR B1 SUREXPRIME
Comme nous l’avons détaillé plus haut, les souris invalidées pour le RB2 surexpriment le RB1. Bien que cette surexpression ait été rapportée par plusieurs autres groupes, les conséquences fonctionnelles et physiopathologiques de ces observations n’ont jamais été envisagées. Les conséquences observées chez les animaux KOB2 pourraient donc ne pas résulter uniquement de l’absence du RB2 mais également de la surexpression du RB1. L’expression du RB1 est toujours associée à des situations de stress, et n’a jamais été rapportée en situation physiologique. Le modèle de la souris invalidée pour le RB2 permet l’étude des effets de l’expression spontanée du RB1 en situation physiologique en absence de stress.
Afin d’étudier l’implication potentielle de la surexpression du RB1 sur la fonction rénale chez les souris KOB2, nous avons exploré les fonctions rénales de 5 groupes de souris : 1) souris C57 Bl6 sauvages (WT) ; 2) souris C57 Bl6 sauvages traitées par un antagoniste du RB2 (WT HOE) ; 3) souris C57 Bl6 invalidées pour le RB2 (KOB2) et 4) souris KOB2 traitées par un antagoniste du RB1. Les animaux du groupe 2 ont reçu une injection sous-cutanée quotidienne de 250 µg/kg/jour d’HOE-140 pendant les 4 jours précédent l’exploration des fonctions rénales. L’antagoniste du RB1 (SSR2406120 Sanofi-Aventis) a été administré quotidiennement par gavage, à la dose de 10 mg/kg pendant les 4 jours précédent l’exploration des fonctions rénales.
• Phénotype des animaux (Figure 2)
Figure 2 : Phénotype des animaux
PSA : Pression sanguine artérielle ; WT : souris sauvages ; Antag B2 : antagoniste du RB2 (HOE-140) ; Antag B1 antagoniste du RB1 (SSR2406120) ; B2KO : souris invalidées pour le RB2.
L’invalidation ou le blocage pharmacologique du RB2 n’ont pas d’effets significatifs sur le poids corporel, le poids des reins, la glycémie et la pression sanguine artérielle. La glycémie tend à être légèrement réduite chez les souris KOB2 et réaugmentée en présence de l’antagoniste du RB1, mais ce résultat n’est pas significatif.
WT WT An tag B2 B2K O B2K O A nta g B1 0 25 50 75 100 PS A ( m m H g ) WT WT An tag B2 B2K O B2KO An tag B1 0.0 0.5 1.0 1.5 2.0 2.5 G ly cém ie ( g /L ) WT WT A ntag B2 B2KO B2KO Anta g B 1 0 5 10 15 20 25 30 35 P o id s co rp o rel ( g ) 0.30 0.25 0.20 0.15 0.10 0.05 0.35 P o id s d e s r e in s ( g ) 0.00 WT WT An tag B2 B2K O B2K O An tag B 1
• Effets sur les paramètres fonctionnels rénaux (Figure 3)
Figure 3 : Paramètres fonctionnels rénaux
DFG : Débit de Filtration Glomérulaire ; DPR : Débit Plasmatique Rénal ; FF : Fraction Filtrée ; RVR : Résistances Vasculaires Rénales.
L’invalidation du RB2 a pour principale conséquence une réduction du Débit de Filtration Glomérulaire (DFG), avec un Débit Plasmatique Rénal inchangé (DPR), et par conséquent une réduction de la Fraction Filtrée (FF). Ces effets ne sont pas observés chez les souris traitées par l’antagoniste du RB2 (qui n’expriment pas le B1), et sont annulés chez les souris KOB2 traitées par l’antagoniste du RB1, suggérant qu’ils résultent de la surexpression du RB1.
Le même travail est en cours chez les animaux invalidés pour le RB1 et traités ou non par un antagoniste du RB2. Deux groupes supplémentaires de souris sauvages sont en cours d’étude : l’un traité par l’antagoniste du RB1, et l’autre traité simultanément par les deux antagonistes (RB1 et RB2). WT WT A ntag B2 B2 KO B2K O An tag B 1 0 100 200 300 * * D F G ( µ l/m in /6 5 c m 2) 1500 1000 500 * D P R (µl/m in /6 5 c m 2) 0 WT WT An tag B 2 B2KO B2K O A ntag B1 ** 35 WT WT An tag B2 B2K O B2KO An tag B1 0 5 10 15 20 25 30 * *** FF ( % ) 75 50 25 0 100 RV R ( m m Hg/ m l/ m in /c m 2) WT WT An tag B2 B2KO B2KO An tag B1
III.4. PLACE DE LA BRADYKININE DANS LES EFFETS DU BLOCAGE DU SYSTEME RENINE-ANGIOTENSINE
Les premiers arguments en faveur du rôle néphroprotecteur de la BK sont indirects et reposent sur les effets bénéfiques du blocage du système rénine-angiotensine au cours des néphropathies hypertensives et diabétiques. Ces deux néphropathies se caractérisent par une atteinte glomérulaire précoce avec accumulation de matrice mésangiale, une glomérulosclérose progressive et des lésions de fibrose tubulo-interstitielle qui évoluent vers l’IRC. En effet, ces pathologies sont souvent associées à des complications au niveau rénal, caractérisées par une hyperplasie, une hypertrophie cellulaire et une accumulation de matrice extracellulaire initialement localisée dans le glomérule. Elles aboutissent à la fibrose du glomérule (ou glomérulosclérose), à la fibrose interstitielle et à l’insuffisance rénale. Actuellement, le blocage du système rénine-angiotensine (SRA) par les inhibiteurs de l’enzyme de conversion (IEC) ou par les antagonistes du récepteur AT1 de l’angiotensine II (ARA2) s’avère la meilleure stratégie thérapeutique [134] pour ralentir l’évolution et la sévérité de la glomérulosclérose.
III.4.a. Inhibiteurs de l’Enzyme de Conversion
L’effet protecteur cardiovasculaire et rénal des IEC a été démontré dans plusieurs grandes études cliniques. Par exemple, l’étude REIN (The Ramipril Efficacy In Nephropathy, [135] a montré que le traitement de patients protéinuriques par un IEC (ramipril), permet malgré une pression artérielle identique, de ralentir le déclin du débit de filtration glomérulaire, et de réduire le risque de développer une insuffisance rénale terminale. L’effet protecteur rénal des IEC serait supérieur à celui des autres molécules anti-hypertensives. L’étude HOPE (Heart Outcomes Prevention Evaluation [136]) a montré que le traitement par
le ramipril est associé à une réduction de la mortalité et de la morbidité cardio-vasculaire chez les patients à haut risque cardio-vasculaire. Ce bénéfice est encore plus grand chez les patients présentant des pathologies rénales, ce qui suggère un effet spécifique des IEC sur la protéinurie, indépendamment de leur effet hypotenseur [137].
Les IEC agissent à la fois sur le SRA vasoconstricteur et le SKK vasodilatateur. Leurs effets bénéfiques ont longtemps été rapportés uniquement à l’inhibition du système rénine-angiotensine. Pourtant, actuellement de plus en plus d’arguments suggèrent que l’accumulation de BK joue un rôle aussi important. Tout d’abord, la biodisponibilité d’angiotensine II n’est pas toujours modifiée lors d’un traitement par des IEC [138, 139], probablement en raison de la possibilité d’une synthèse d’angiotensine II indépendante de l’ECA, par exemple via les chymases [140]. La concentration d’angiotensine II est également dépendante de sa dégradation par l’enzyme de conversion 2 de l’angiotensine II (ECA 2). Ce résultat suggère que le blocage de la synthèse d’angiotensine II n’est pas à lui seul responsable de tous les effets des IEC. L’utilisation d’antagonistes du récepteur B2 a permis d’apporter des preuves expérimentales en faveur du rôle de ce récepteur dans l’action protectrice des IEC. Les effets bénéfiques des IEC sont en effet souvent atténués par le blocage du RB2. Les IEC exercent leur effet anti-fibrosant d’une part en inhibant la synthèse de composants de la matrice extracellulaire (collagène et fibronectine) et d’autre part en stimulant sa dégradation (via l’inhibition de l’expression de la protéine PAI-1 (Plasminogen Activator Inhibitor-1). Ces deux actions des IEC sont supprimées par l’administration d’un antagoniste du RB2 [86, 141]. L’effet anti-fibrosant des IEC est donc largement dépendant de la bradykinine et de la mise en jeu de son récepteur B2. De la même façon, l’effet anti-protéinurique des IEC au cours de néphropathies expérimentales chez le rat, est en partie dépendant des kinines [142, 143].
L’activation du RB2 par les IEC peut se faire par plusieurs mécanismes intriqués et additionnels (Figure 4). Le premier d’entre eux est, bien sûr, l’inhibition de la dégradation de la BK, mais de nouveaux mécanismes ont été proposés. Tout d’abord, l’enzyme de conversion de l’angiotensine (ECA) et le RB2 pourraient former un complexe (complexe ECA/RB2), qui augmenterait la sensibilité du RB2 pour la bradykinine [144]. Par ailleurs, l’inhibition de l’ECA entraîne l’accumulation d’angiotensine I, qui est dégradée en angiotensine 1-7 par plusieurs enzymes dont l’ECA2 (Figure 4). L’angiotensine 1-7 agit en activant un récepteur spécifique encore imparfaitement identifié [145, 146], mais qui pourrait être associé à l’oncogène Mas [147]. Les effets de l’angiotensine 1-7, hypotenseurs et vasodilatateurs, sont opposés à ceux de l’angiotensine II. Par ailleurs, certains de ces effets sont inhibés par les antagonistes du RB2 [148, 149]. L’angiotensine 1-7, peut se comporter comme un inhibiteur de l’ECA et donc augmenter la biodisponibilité de la BK mais également agir sur le complexe ECA/RB2 et diminuer la désensibilisation du RB2 [150]. Bien que tous les mécanismes d’action de l’angiotensine 1-7 ne soient pas encore identifiés, les voies de formation associées à la stimulation potentielle du RB2 peuvent permettre de définir de nouvelles voies thérapeutiques.