THESE
En vue de l’obtention du
DOCTORAT DE L’UNIVERSITE DE TOULOUSE
Délivré par l’Université Toulouse III – Paul Sabatier
Discipline ou Spécialité Neurosciences
Présentée et soutenue par Caroline ANDRE Le 2 octobre 2008
Titre Etude du rôle des cytokines dans l’activation de l’indoléamine 2,3-dioxygénase cérébrale impliquée dans les altérations comportementales
associées à l’inflammation
Jury
Bernard FRANCES, Professeur: Président Odile VILTART, Chargée de recherche: Rapporteur Pascal CLAYETTE, Directeur de recherche: Rapporteur Jean FIORAMONTI, Directeur de recherche: Examinateur Robert DANTZER, Directeur de recherche: Examinateur
Nathalie CASTANON, Chargée de recherche: Examinateur
Ecole doctorale : CLESCO
Unité de recherche : Laboratoire de Psychoneuroimmunologie, Nutrition et Génétique Directeur (s) de thèse : Nathalie Castanon
Un grand grand merci !!
Je tiens à remercier Monsieur Bernard Frances de m’avoir fait l’honneur d’accepter de présider le jury de cette thèse.
Je remercie sincèrement Madame Odile Viltart et Monsieur Pascal Clayette d’avoir accepté d’être rapporteurs et de prendre le temps malgré tout de juger ce travail de thèse.
Je tiens également à remercier Monsieur Jean Fioramonti d’avoir accepté de participer à ce jury. Je remercie tout particulièrement Monsieur Robert Dantzer pour m’avoir fait confiance et m’avoir permis de réaliser une partie de ma thèse dans le grand froid de l’Illinois. Ce fut une expérience très enrichissante sur bien des points. Encore merci!
Nathalie, trouve ici toute ma reconnaissance pour m’avoir soutenue et encouragée tout au long de ce très long parcours parsemé d’embûches. Merci de t’être autant investie et d’avoir tenu bon malgré tout. Finalement, on y est arrivées !!
I would like to sincerely thank Professor Keith Kelley for welcoming me in his laboratory. I would also thank all the “American IDO team”, especially Jason and Marc for your helpfulness and your unremitting jokes. I will never forget these months spent in your lab. It was really a great experiment. Thank you!!
Je remercie sincèrement Françoise Moos pour m’avoir accueilli au sein de son laboratoire.
A mes « grandes sœurs de thèse » : Agnès, mon « modèle », merci pour ta confiance (tu en as pour nous deux !) et pour ton soutien depuis le début de cette aventure. Merci d’avoir insisté à l’époque, ça en valait le coup !! Je te souhaite plein de bonheur avec l’arrivée de ton ptit bout.
Maïté, toi qui m’a guidé au travers du vaste univers de l’IDO. C’était sympa de pouvoir partager avec toi les questions existentielles « du pourquoi du comment ». Je te souhaite plein de bonnes choses pour la suite mais je sais déjà que ça en prend bien le chemin.
Ces longs mois loin de chez nous n’ont pas toujours été super évident mais j’en garde plein de bons souvenirs grâce à toi.
Merci à tous les membres du laboratoire Psynugen pour m’avoir aidé, chacun à sa façon, à l’élaboration de ce travail.
Un petit clin d’œil aux inconditionnelles de la « cafèt » du midi avec qui il était bien sympa de décompresser entre deux paragraphes.
Jérôme, merci pour ton soutien et d’avoir été aux petits soins pendant cette longue et stressante période d’écriture. Le meilleur est à venir.
Papa et Maman, vous qui avez la lourde tâche de me supporter depuis tant d’années… Merci pour votre soutien inconditionnel, tout ça c’est grâce à vous. Merci d’être aussi forts et d’être toujours là pour moi malgré tout.
Table des matières
Table des matières
3Résumé
8Abstract
10Introduction
12Avant propos 12
La réponse immunitaire 14
I- Rôle des cytokines dans la réponse inflammatoire 14
1- Réponse inflammatoire innée 14
2- Les cytokines 15
a- Le TNFα 16
b- L’IFNγ 18
II- Rôles des cytokines dans la réponse systémique et centrale 20
1- Composante systémique et centrale 20
2- Communication système immunitaire/ système nerveux 21
3- Conséquences de l’action régulée des cytokines sur le cerveau 22
Développement des troubles de l’humeur : conséquences centrales de l’action
dérégulée des cytokines 23
I- Description de la symptomatologie de la dépression 23
II- Etiologie de la dépression 24
1- Théorie neuroendocrinienne, neurochimique et anatomique 24
2- Théorie immunitaire 25
Cytokines et altération du métabolisme du tryptophane : implication
potentielle dans le développement des troubles de l’humeur 27
I- Rôle du tryptophane dans le métabolisme de la sérotonine 27
1- Rôle et distribution de la sérotonine 27
2- Biosynthèse et métabolisme 27
3- Modulation du métabolisme de la sérotonine 28
II- Implication de la voie de la kynurénine dans la modulation de la synthèse de
sérotonine 30
1- Les enzymes : TDO et IDO 30
2- Biochimie, génétique et régulation de l’IDO 31
Table des matières
a- Rôle antiprolifératif 32
b- Rôle immunosuppresseur au cours de la gestation 33 c- Rôle immunosuppresseur lors d’un rejet de greffe 34 d- Rôle immunosuppresseur lors du développement de tumeurs 35
III- Rôle de l’IDO dans le développement des troubles de l’humeur 36
1- Implication potentielle de l’IDO dans les altérations neurochimiques de la
dépression : hypothèse sérotoninergique 36
2- Implication potentielle de l’IDO dans les altérations neuronales de la
dépression : hypothèse glutamatergique 38
Implications physiopathologiques de l’altération de la régulation des
cytokines : cas de l’obésité 39
I- Obésité : une pathologie inflammatoire chronique à bas bruit 39
1- Prévalence de l’obésité 39
2- Physiologie de l’obésité 40
3- Origine de l’inflammation 41
a- Leptine et résistine 42
b- TNFα et IL-6 44
II- Rôle potentiel de l’IDO dans le développement des troubles de l’humeur
associés à l’obésité 47
1- La relation entre l’obésité et la dépression 47
2- Bases biologiques de la relation entre obésité et dépression 48
Objectifs
51Démarche expérimentale
541ère approche : Etude de l’implication de l’IDO dans la mise en place des
troubles de l’humeur observés chez la souris lors d’une stimulation aiguë du système immunitaire.
54
1- Le LPS : un modèle d’activation aiguë du système immunitaire 54
2- Démarche expérimentale 55
2ème approche : Etude de l’implication de l’IDO dans la mise en place des
troubles de l’humeur lors d’une stimulation chronique du système immunitaire. 56
1- Le BCG : un modèle d’activation chronique du système immunitaire 57
2- Démarche expérimentale 58
3ème approche : Etude des conséquences de l’inflammation liée à l’obésité sur
l’activation de l’IDO et des troubles comportementaux associés. 59
1- Régime enrichi entraînant l’obésité: un modèle physiopathologique
d’inflammation chronique. 59
2- Démarche expérimentale 60
Résultats
63Chapitre 1 64
Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice
Chapitre 2 79
Spatio-temporal differences in the profil of murine brain expression of proinflammatory cytokines and indoleamine 2,3-dioxygenase in response to peripheral lipopolysaccharide administration
Chapitre 3 92
Inoculation of Bacillus Calmette-Guerin to mice induces an acute episode of sickness behavior followed by chronic depressive-like behavior
Chapitre 4 104
Interferon-γ is required for activation of indoleamine 2,3-dioxygenase and depressive-like behavior induced by Bacillus Calmette-Guerin
Chapitre 5 118
Diet-induced obesity in mice exacerbates neuroinflammation and sickness elicited by systemic infection
Synthèse des principaux résultats
155Discussion générale
157IDO : molécule clé à l’interface entre SI et troubles de l’humeur 157
I- Dissociation comportement de maladie / troubles de l’humeur 158
1- Modèle d’activation aiguë du SI 158
2- Modèle d’activation chronique du SI 160
II- Implication de l’IDO dans la mise en place des troubles de l’humeur 163
1- Conséquences du blocage direct de l’IDO 163
2- Conséquences du blocage de la production de cytokines inflammatoires 165
Mécanismes de l’induction de l’IDO lors d’une stimulation immune aiguë du
SI 168
I- Expression centrale de l’IDO 168
II- Rôle prépondérant de l’IFNγ ? 170
III- Action synergique des cytokines inflammatoires ? 172
Mécanismes de l’induction de l’IDO lors d’une stimulation immune chronique
du SI 176
I- Expression centrale de l’IDO 176
II- Rôle prépondérant de l’IFNγ ? 177
III- Importance du TNFα 179
Conséquences de l’obésité sur la réponse à l’infection 182
Table des matières
II- Conséquences sur la réponse inflammatoire 184
III- Vulnérabilité à une infection 186
1- Du système immunitaire 186
2- De l’activation de l’IDO 188
3- De l’activation de l’axe HPA 190
4- De la réactivité comportementale 191
Conclusion et perspectives
195Bibliographie
201Abréviations
244Index des figures
247Abstract
248Résumé et abstract
Résumé
Lors d’une infection, les cytokines inflammatoires produites localement par les cellules de l’immunité innée activées sont capables, grâce à leurs propriétés endocrines, d’induire la production de ces mêmes signaux moléculaires par la microglie et les macrophages résidents du système nerveux central (SNC). Cette image en miroir de la réponse périphérique aux infections organise les composantes subjectives, comportementales et métaboliques du comportement de maladie qui s’accompagne d’une hyperactivité de l’axe hypothalamo-hypophyso-corticosurrénalien (HPA), ainsi que de l’altération des systèmes monoaminergiques centraux. Cette réponse adaptative transitoire et réversible de l’hôte permet à l’organisme de lutter de façon appropriée contre l’infection et de maintenir son homéostasie. Cependant, il a récemment été montré chez l’animal que ces symptômes de maladie peuvent être suivis de l’apparition de symptômes de type dépressif lorsque la stimulation du système immunitaire (SI) se maintient de façon incontrôlée. De plus, des études cliniques montrent que l’administration thérapeutique chronique de cytokines inflammatoires induit la mise en place de troubles de l’humeur. Une forte prévalence de ces troubles est également observée dans un grand nombre de pathologies à composante inflammatoire, telle que l’obésité, étayant l’hypothèse immunitaire de la dépression.
L’intensité des troubles de l’humeur observés chez les patients traités par immunothérapie est corrélée à une diminution des taux plasmatiques de tryptophane (Trp), un acide aminé essentiel servant de précurseur et de facteur limitant à la synthèse de sérotonine. Or, nous avons montré au laboratoire que l’activité de l’indoléamine 2,3-dioxygénase (IDO), l’enzyme initiant le catabolisme du Trp vers la voie de la kynurénine et de ses dérivés neurotoxiques, était fortement augmentée en périphérie ainsi qu’au niveau cérébral lors d’une stimulation aiguë ou chronique du SI. Ainsi, l’objectif majeur de ce travail de thèse a été de déterminer chez la souris, le rôle potentiel de l’IDO dans la mise en place de troubles de l’humeur induits en conditions inflammatoires et d’identifier les mécanismes sous-tendant son activation au niveau cérébral.
Nos résultats montrent que : (1) l’IDO est directement impliquée dans le développement des troubles de l’humeur puisque son inhibition pharmacologique prévient la mise en place des symptômes de type dépressif induit lors d’une stimulation aiguë du SI par le lipopolysaccharide (LPS); (2) l’activation des mécanismes intracellulaires sous-tendant l’expression cérébrale de l’IDO induite par le LPS est fortement conditionnée par la
disponibilité locale de l’interféron-γ (IFNγ); (3) l’IFNγ et en moindre mesure, le facteur de nécrose tumorale (TNFα) participent au développement soutenu des changements comportementaux de type dépressif associés à l’activation cérébrale prolongée de l’IDO induite par une stimulation chronique du SI par le bacille de Calmette et Guérin (BCG); (4) les modifications physiopathologiques induites par le développement d’un état d’obésité altèrent la capacité du système immunitaire à répondre de manière adaptée à une infection, comme en témoigne l’activation accrue de la production de cytokines périphériques et cérébrales, en particulier l’interleukine-6 (IL-6), l’exacerbation des réponses neurochimiques (stimulation de l’IDO pulmonaire et cérébrale) et neuroendocriniennes (sur-activation de l’axe HPA) et de leurs conséquences sur la réactivité comportementale.
En conclusion, ces résultats originaux permettent de mettre en évidence le rôle clé de l’IDO dans l’apparition de troubles de l’humeur associés à l’inflammation et de commencer à préciser les mécanismes sous-tendant son activation cérébrale in vivo. Ce travail pourrait ainsi contribuer à l’identification de cibles moléculaires potentielles pour le développement de nouvelles stratégies thérapeutiques visant à améliorer la qualité de vie des patients atteints de pathologies à composante inflammatoire associées à une symptomatologie dépressive.
Résumé et abstract
Study of the role of cytokines in the activation of brain indoleamine
2,3-dioxygenase (IDO) involved in behavioral and mood alterations associated
with inflammation
Proinflammatory cytokines transiently expressed by glial cells in the brain during an infection organize the physiological and behavioral components of sickness but can also induce mood alterations, as shown in cancer patients chronically treated with cytokines. The occurrence of mood disorders is associated with a drastic fall in plasma levels of tryptophan, the amino-acid precursor and limiting factor of serotonin synthesis. This drop could be explained by the activation of the indoleamine 2,3-dioxygenase (IDO), a tryptophan-catabolizing enzyme potently induced in monocytes and brain microglia by cytokines.
The present study aimed therefore at determining whether brain IDO activation is required for cytokine-induced depressive-like behavior development and to identify the mechanisms underlying brain IDO induction in inflammatory conditions.
Our results demonstrate for the first time that 1) IDO is a critical mediator of depressive-like behavior in response to acute stimulation of the innate immune system induced by lipopolysaccharide (LPS); 2) activation of intracellular mechanisms underlying LPS-induced IDO expression in the brain is mediated by local availability of IFNγ; 3) IFNγ and TNFα participate in brain IDO activation and induction of depressive-like behavior in mice following Bacillus Calmette-Guerin (BCG)-induced chronic inflammation, although the role played by IFNγ might likely be predominant; 4) diet-induced obesity interferes with the ability of the immune system to appropriately respond to bacterial LPS challenge.
Taken together, these findings constitute a first important step towards the identification of new pharmacological strategies aimed at preventing mood alterations associated with diseases that share a common inflammatory component.
Introduction
Introduction
Avant-propos
Le système nerveux central (SNC) a longtemps été considéré comme un organe immunologiquement privilégié, complètement protégé des cellules immunitaires circulantes par la présence de la barrière hématoencéphalique (BHE) et présentant une sensibilité immune altérée voire inexistante. Cependant, ce concept a été abandonné avec la mise en évidence de nombreuses interactions entre le SNC, le système immunitaire et le système neuroendocrinien (Ader et al., 1995). Ainsi lors d’une infection, les cellules immunitaires sont capables de traverser la BHE pour s’infiltrer dans le parenchyme cérébral et induire la réponse inflammatoire centrale via la production et la libération de cytokines. La démonstration d’une communication bidirectionnelle entre SNC et système immunitaire a suggéré qu’en plus de son rôle dans la défense de l’organisme face aux infections, le SI pourrait être impliqué dans le développement de processus neuropathologiques et plus particulièrement dans l’apparition de troubles psychiatriques tels que la dépression (Miller et al., 1999). Un grand nombre de pathologies comprenant une composante inflammatoire telles que l’athérosclérose, le syndrome d’immunodéficience acquise (SIDA), certains types de cancers, l’arthrite rhumatoïde, les maladies coronariennes ou encore l’obésité, est associé à une forte prévalence des troubles de l’humeur (Evans et al., 2005). De plus, de telles altérations sont également fréquemment observées lors de l’administration thérapeutique chronique de cytokines inflammatoires chez des patients atteints de cancers ou encore d’une hépatite C (Bonaccorso et al., 2002; Capuron et al., 2002), étayant ainsi la théorie d’une composante immunitaire dans le développement de troubles de l’humeur. Le système immunitaire et plus particulièrement les cytokines jouent donc un rôle important dans l’apparition des troubles de l’humeur associés à un état inflammatoire. L’identification de marqueurs biologiques de l’inflammation impliqués dans la mise en place de ces troubles et représentant des cibles thérapeutiques potentielles serait donc un atout majeur pour aider au développement de nouvelles stratégies visant à prévenir l’apparition des troubles de l’humeur et améliorer ainsi la qualité de vie des patients.
Chez les patients traités par immunothérapie, l’intensité des symptômes dépressifs observés a pu être corrélée positivement avec la diminution des concentrations plasmatiques
de tryptophane (Trp) (Capuron et al., 2002), un acide aminé essentiel provenant uniquement de la dégradation des protéines de l’alimentation. Dans l’obésité, maladie se caractérisant par un état inflammatoire chronique associé à de nombreuses autres pathologies parmi lesquelles une prévalence élevée de troubles de l’humeur, le développement de ces derniers est proportionnel à l’indice de masse corporelle (Stunkard et al., 2003). De plus, il a pu être observé une réduction des taux circulants de Trp chez les sujets obèses (Breum et al., 2003; Brandacher et al., 2006). Au sein de l’organisme, le Trp, qui participe à la synthèse protéique comme tous les autres acides aminés, est aussi utilisé soit comme précurseur de la synthèse de sérotonine (5-HT), soit catabolisé via la voie de la kynurénine, favorisant alors la formation de composés neurotoxiques agissant sur les récepteurs au glutamate (Wirleitner et al., 2003). Il est désormais connu que les troubles de l’humeur sont entre autre, fortement associés à une altération des systèmes sérotoninergiques et glutamatergiques (Müller et Schwarz, 2007). Ainsi, ces données suggèrent qu’en conditions inflammatoires, la dérégulation du métabolisme du Trp pourrait jouer un rôle causal dans l’apparition des troubles de l’humeur.
Différentes données expérimentales obtenues dans notre laboratoire et complétant celles décrites dans la littérature notamment chez l’homme, nous ont amené à proposer qu’une des interactions entre le SI et le métabolisme du Trp passe par l’activation de l’enzyme initiant le catabolisme du Trp en conditions inflammatoires, l’indoléamine 2,3-dioxygénase (IDO). En effet, il a été montré au laboratoire que l’activité enzymatique de l’IDO est fortement induite en périphérie ainsi qu’au niveau cérébral après une stimulation aiguë ou chronique du SI chez la souris (Lestage et al., 2002; Moreau et al., 2005) et que cette activation est associée à l’apparition de changements comportementaux de type dépressif (Frenois et al., 2007). Cependant, le rôle exact de l’IDO dans ce phénomène, ainsi que les mécanismes à l’origine de son activation au niveau cérébral sont encore mal connus. Ces données sont cependant cruciales pour le développement de nouvelles stratégies thérapeutiques adaptées. Dans ce contexte, ce travail de thèse a visé à identifier les mécanismes sous-tendant l’activation de l’IDO cérébrale et le développement de troubles de l’humeur en conditions inflammatoires d’origine infectieuse ou liées à une pathologie à composante inflammatoire telle que l’obésité.
Introduction
La réponse immunitaire
La réponse de l’organisme à une infection se caractérise par l’activation des deux composantes du système immunitaire : l’immunité innée ou non spécifique et l’immunité
spécifique ou adaptative qui agissent en synergie afin d’enrayer la progression et le
développement des agents pathogènes et maintenir ainsi l’intégrité de l’organisme. L’immunité innée est considérée comme la première ligne de défense de l’organisme car elle est activée dès l’infiltration d’un agent pathogène et permet son élimination par le biais des cellules phagocytaires présentes localement. Son action se décline en trois niveaux :
- la composante locale, également appelée réponse inflammatoire - la composante systémique
- la composante centrale.
I- Rôle des cytokines dans la réponse inflammatoire
1- Réponse inflammatoire innée
La réponse inflammatoire se caractérise par un ensemble de perturbations physiologiques locales, immédiates et transitoires. Elle se traduit par les 4 signes cardinaux de l’inflammation décrits dès le premier siècle avant J.C. par Cornelius Celsus : « rubor et tumor cum calor et dolor » (rougeur, tuméfaction avec chaleur, douleur). Puis, Claude Galien (131-201 après J.C.) ajoutera un 5ème point cardinal « functio laesa » (perte de fonction) (Rather, 1982). Ces signes cliniques témoignent d’une augmentation de la perméabilité des vaisseaux sanguins facilitant la diapédèse des leucocytes circulants vers le site de l’infection et d’une vasodilatation ayant pour but d’augmenter le flux sanguin et de permettre l’apport rapide de protéines. L’exsudation du plasma qui en résulte provoque un gonflement des tissus qui, en comprimant les terminaisons nerveuses, est responsable de la douleur. Ces modifications s’accompagnent également de l’attraction par chimiotactisme et l’accumulation des cellules de l’immunité innée telles que les monocytes, macrophages ou encore neutrophiles. Ces cellules sont capables de se lier aux molécules antigéniques présentes à la surface des agents infectieux pour induire leur phagocytose et produisent des molécules inflammatoires telles que les cytokines permettant le recrutement d’un grand nombre de cellules de l’immunité innée au site de l’infection (Figure 1).
Introduction
2- Les cytokines
Les cytokines jouent un rôle complexe mais central dans l’induction de la réponse inflammatoire. Polypeptides de poids moléculaire moyen (entre 8 et 30 KDa), elles sont produites par un grand nombre de types cellulaires différents incluant des cellules immunitaires (macrophages et monocytes) ou non immunitaires (cellules endothéliales et fibroblastes) et chaque cellule donnée produit le plus souvent plusieurs cytokines distinctes. De plus, les cytokines agissent en cascade et forment ainsi un réseau inter-dépendant de médiateurs qui modulent leur propre synthèse, ainsi que celle des autres cytokines du réseau par des boucles de rétroaction positives et négatives (Cavaillon et Haeffner-Cavaillon, 1993). Leur action est dite pléiotrope (elles agissent sur plusieurs types de cibles cellulaires) et redondante (plusieurs cytokines peuvent avoir les mêmes effets sur une même cellule). Elles se fixent à des récepteurs membranaires spécifiques dont l’expression est souvent régulée par les cytokines elles-mêmes et ont une fonction autocrine (sur la cellule productrice), juxtacrine (sur des cellules en contact), paracrine (sur des cellules proches) ou encore endocrine (sur des cellules distantes, via la circulation sanguine).
Au cours de la réponse inflammatoire, les cytokines exercent des activités biologiques multiples. Elles régulent notamment la synthèse des molécules d’adhésion par les cellules endothéliales facilitant la diapédèse des leucocytes vers les tissus et leur accumulation au site inflammatoire (May et Ager, 1992). Les cytokines induisent également la production de prostaglandines, molécules impliquées entre autre, dans l’équilibre vasodilatation/constriction des vaisseaux. Libérés par les fibroblastes ainsi que les macrophages, ces médiateurs jouent un rôle important dans les premiers stades de l’inflammation en augmentant la perméabilité vasculaire. Localement, les cytokines permettent la production de radicaux libres par les cellules endothéliales. Néanmoins, si ces composés contribuent à l’activité microbicide, ils sont également toxiques pour les tissus environnants (Klebanoff, 1992).
Les cytokines constituent une famille hétérogène de molécules qui peut cependant être divisé en deux grands groupes : les cytokines inflammatoires et les anti-inflammatoires. Le premier groupe rassemble principalement les cytokines responsables de la réponse inflammatoire dont les interleukines IL-1β, IL-6 et IL-12, le facteur de nécrose tumorale
TNFα et les interférons, principalement l’IFNγ. Le second groupe rassemble les cytokines
régulant cette réponse en inhibant la production et l’action des cytokines inflammatoires. Il réunit l’IL-10, l’antagoniste du récepteur de l’IL-1 (IL-1Ra), l’IL-4, l’IL-13 et le TGFβ (transforming growth factor beta). Les trois cytokines inflammatoires les plus étudiées et
largement impliquées dans la mise en place de la réponse inflammatoire locale, systémique et centrale sont l’IL-1β, l’IL-6 et le TNFα (Baumann et Gauldie, 1994). Lors d’une infection aiguë, l’immunité innée activée met en jeu une réponse inflammatoire non spécifique et induit une production massive mais transitoire de cytokines, ne dépassant pas 24 à 48 h. Cependant, cette réponse inflammatoire peut devenir chronique lorsque le système immunitaire ne parvient pas à éliminer le pathogène. La réponse immunitaire spécifique ou réponse adaptative alors mise en place met en jeu les macrophages qui libèrent l’IL-12, recrutant les lymphocytes T de type helper et les cellules natural killer (NK) (Brunda, 1994). Ces dernières activées sécrètent l’IFNγ provoquant la différenciation des lymphocytes T vers un phénotype Th1. La sous- population Th1 induit une réponse immune à médiation cellulaire (phagocytose) en agissant comme activateur des macrophages au travers d’une forte production d’IFNγ (Janeway et Travers, 1997). La réponse de type Th2, à médiation humorale (anticorps), permet d’induire la production d’IL-4 et d’IL-10 agissant notamment comme régulateur de la réponse Th1 (Tada et al., 1978; Mosmann et al., 1986) (Figure 2). L’inflammation chronique peut alors durer plusieurs semaines et dans certains cas des années (tuberculose, polyarthrite rhumatoïde…).
a- Le TNFα
Le TNF a été identifié dès 1975 grâce à sa capacité à éliminer les cellules tumorales chez la souris (Carswell et al., 1975). Parallèlement, un facteur appelé la cachectine a été isolé à partir de macrophages murins, séquencé et identifié comme étant identique au TNF (Beutler et al., 1985a). La cachectine, décrite comme une hormone catabolique, était connue pour inhiber l’expression d’enzymes anaboliques du tissu adipeux (Kawakami et al., 1982; Pekala et al., 1984; Torti et al., 1985). Puis, d’autres études ont prouvé les puissants effets inflammatoires du TNF, ainsi que son rôle comme médiateur endogène central du choc endotoxique (Dayer et al., 1985; Gamble et al., 1985; Beutler et al., 1985b). La famille du TNF comporte deux cytokines: le TNFα principalement produit par les macrophages et le TNFβ ou lymphotoxine α exclusivement induit par les lymphocytes et les cellules NK, ainsi qu’une protéine soluble de liaison au TNF, le TNF binding protein (TNFbp) (Carswell et al., 1975; Shacks et al., 1973). Le TNFbp a un poids moléculaire de 30 KDa et dérive du domaine de liaison du TNF présent sur le récepteur membranaire (Nophar et al., 1990). Cette protéine régule l’activité du TNF en inhibant sa liaison à son récepteur (Engelmann et al., 1989).
Le gène du TNFα est situé sur le chromosome 17 chez la souris et code pour une protéine précurseur de 26 KDa comportant 235 acides aminés (Nedospasov et al., 1986;
IL-6 IL-10 Fibroblaste Macrophage Cellule endothéliale Cellule NK Lymphocyte B Lymphocyte Th IL-1 IL-6 TNFα IL-10 IL-1 TGFβ PDGF TNF IFNα TGFβ TNF TNFα IL-1 TGFβ IL-12 Lymphocyte Th1 Lymphocyte Th2 IL-2 IL-2 IL-4 IL-10 IFN IL-4 IL-10 TGFβ IFNγ IL-2 TNFα Lymphocyte Tc IL-4 IFNγ IL-12 IL-2 IL-1 IL-2 IFN
Figure 2: Le réseau des cytokines
IFNα: Interféron alpha, IFNγ: Interféron gamma, IL: interleukine, Lymphocyte Tc: Lymphocyte T cytotoxique, Lymphocyte Th: Lymphocyte T helpher, NK: Natural killer, PDGF: Platelet derived growth factor, TGFβ: Transforming growth factor beta,
Pennica et al., 1985). Puis une métalloprotéase, la TNFα converting enzyme clive le proTNF en forme mature de 17 KDa comportant 157 acides aminés (Moss et al., 1997; Jue et al., 1990). Le TNFα se fixe sur son récepteur sous forme d’un trimère composé de trois sous unités de 17 KDa chacune (Jones et al., 1989) (Figure 3).
Il existe deux isoformes du récepteur au TNF liant à la fois le TNFα et le TNFβ, le TNFRI (55KDa) et le TFNRII (75KDa) (pour revue Bazzoni et Beutler, 1996; Bradley, 2008). De nombreuses études ont révélé que le TNFRI induit la majorité des activités biologiques du TNF. Ainsi, il joue un rôle critique dans la réponse inflammatoire et l’apoptose car il possède dans sa partie C-terminale un domaine appelé « Death Domain » (DD) impliqué dans l’activation des voies de signalisation menant à l’apoptose (Chen et al., 2002). La fixation du TNF sur les TNFR entraîne leur trimérisation, ainsi qu’un changement de conformation qui permet la liaison de la protéine adaptatrice TRADD (TNF receptor associated with death domain) au DD. Ce complexe sert de point de départ à l’activation des 3 voies de signalisation induites par le TNFα, la voie conduisant à l’apoptose, la voie NFκB (Nuclear factor kappa B) et la voie des MAPKinases (Mitogen activated protein kinase). Ainsi, l’activation de TRADD induit la liaison de FADD (Fas associated death domain) au niveau de son DD, qui recrute à son tour les caspases dont la caspase-8, impliquées dans l’apoptose (Dempsey et al., 2003).
L’activation de la voie NFκB est initiée par la fixation de la protéine adaptatrice RIP (Receptor interactive protein) à TRADD (Kelliher et al., 1998). Ceci entraîne la phosphorylation des kinases NIK (NFκB Inducing Kinase), IKKα (Inhibiting IκB kinase), IKKβ et NEMO (NFκB Essential Modifier). Ce complexe de kinases activées phosphoryle l’inhibiteur de NFκB, IκB (Inhibitory kappa B) qui se détache de NFκB pour être dégradé par ubiquitination. NFκB, formé par le dimère p50-65, peut alors être transloqué vers le noyau pour induire la transcription des gènes cibles notamment impliqués dans la réponse inflammatoire.
TRADD peut également recruter TRAF2 (TNF receptor associated factor) qui induit l’activation de la voie des MAPKinases. La cascade des MAPKinases se compose de 3 groupes: ERK 1/2 (Extracellular signal regulated kinase 1/2), p38 et JNK (c-Jun N-terminal kinase) (Parker et al., 2002; Chen et al., 2001; Kyriakis et Avurch, 2001). Bien que chaque groupe ait des caractéristiques spécifiques, un certain nombre de critères leur sont communs. De plus, chaque famille de MAPKinases est composée d’un ensemble de 3 kinases: une MAPKKK, une MAPKK et une MAPK. Cette dernière activée catalyse la phosphorylation
TNF α DD TR ADD Apoptose ERK1/
Figure 3: Les voies de signalisation associées au TNFα
FADD RIP Procasp ase 8 TRAF2 ASK1 MEKK1 Casp ase 8 Ras Activation des caspases Raf IKK NIK IKK α β
MEK 1/2 MEK 4/7 MEK 3/6
JNK p38 IκB p50 p65 p50 p65 AP-1 NFκB
AP-1: Activating protein 1, ASK1: Apoptosis signal-regulating kinase 1, DD: Death domain, ERK1/2: Extracellular signal-regulated kinase 1/2, FADD: Fas associated death domain, IκB: Inhibitory κB, IKKα/β: Inhibiting IκB kinase alpha et beta, JNK: c-Jun N-terminal kinase,
MEK: Extracellular signal-regulated kinase kinase, MEKK1: MEK kinase 1, NFκB: Nuclear factor kappa B, NIK: NFκB-inducing kinase, p50: sous-unité du dimère NFκB de 50 KDa, p65: sous-unité du dimère NFκB de 65 KDa, Raf: Sérine/thréonine kinase, Ras: Small GTP binding protein, RIP: Receptor interactive protein, TNFα: Tumor necrosis factor, TRADD: TNF receptor associated with death domain, TRAF2: TNF receptor associated factor 2
des MKs (MAPK-activated protein kinases), permettant ainsi une étape d’amplification supplémentaire. Si ERK 1/2 et JNK sont principalement connus pour être impliqués dans la prolifération et la différenciation cellulaire (Chang et Karin, 2001), p38 semble être essentiel dans les réponses immunitaires et inflammatoires (O’Neill, 2003). En effet, la voie p38 joue un rôle apoptotique puisque son inhibition a un effet neuroprotecteur lors d’une ischémie cérébrale (Barone et al., 2001). De plus, p38 est activé dans les macrophages où il participe à la mise en place de leurs activités fonctionnelles telles que la chimiotaxie, l’exocytose, l’adhérence ou encore l’apoptose (Dong et al., 2002). Il régule également la différenciation et l’apoptose des cellules T en modulant la production d’interférons (Ono et Han, 2000).
b- L’IFNγ
A l’origine, la famille des interférons a été identifiée comme un mécanisme de défense de l’organisme lors d’une infection par le virus de la grippe (Isaacs et Lindenmann, 1957; Isaacs et al., 1957). Les IFNs sont divisés en deux groupes distincts : les IFNs de type I et de type II, agissant via des récepteurs différents. La superfamille des IFNs de type I comporte un grand nombre de sous-types d’IFNα, l’IFNβ, l’IFNω, l’IFNδ et l’IFNτ (trophoblastine) codés chacun par un ou plusieurs gènes (Allen et al., 1996; Roberts et al., 1998). Bien qu’ils soient sécrétés par la plupart des types cellulaires, les leucocytes sont les cellules productrices majoritaires d’IFNα et ω, tandis que l’IFNβ est principalement sécrété par les fibroblastes. L’IFNγ, seul représentant des IFNs de type II et appelé IFN immunitaire, est exclusivement produit par certaines cellules du système immunitaire telles que les lymphocytes T, les cellules NK et les macrophages (Young, 1996). Bien que les IFNs aient tout d’abord été identifiés comme des agents ayant une activité antivirale directe, les propriétés de l’IFNγ incluent également la régulation de certains aspects de la réponse immunitaire, la stimulation de l’activité bactéricide des phagocytes, la régulation des molécules du complexe d’histocompatibilité majeur de classes I et II, l’orchestration des interactions endothélium-leucocytes, ainsi que des effets sur la prolifération cellulaire et l’apoptose (Boehm et al., 1997).
Le gène de l’IFNγ est situé sur le chromosome 10 chez la souris et code pour une protéine de 15,4 KDa comportant 134 acides aminés (Naylor et al., 1984; Gray et Goeddel, 1983). L’IFNγ se fixe sur son récepteur sous forme d’un homodimère de 34 KDa pour activer la voie de signalisation JAK/STAT1 (Janus kinase/Signal of transducer and activator of transcription 1) (Greenlund et al., 1993) (Figure 4). Le récepteur de l’IFNγ se caractérise par deux sous-unités: une chaîne α de 90 KDa (IFNγRI), sous-unité majoritaire et une chaîne β de
IFN γR1 IFN γR2 JAK1 JAK2 P P P P P P STAT STAT P P GAS ISRE STAT P STAT P IRS
Figure 4: La voie de signalisation associée à l’IFNγ
STAT
P
STAT
P
GAS: Gamma activated site, IFNγ: Interféron gamma, IFNγR1 et 2: Interféron gamma récepteur 1 et 2, ISRE: Interferon-stimulated response elements, JAK1 et 2: Janus kinase 1 et 2, STAT1: Signal of transducer and activator of transcription 1
60-67 KDa (IFNγRII), qui permet d’accroître l’affinité du IFNγRI pour son ligand, probablement en augmentant la stabilité du complexe (Marsters et al., 1995). Les chaînes α et β sont constitutivement associées à une Janus kinase spécifique, JAK 1 et 2 respectivement (Igarashi et al., 1994). La fixation de l’IFNγ sur deux IFNγRI entraîne leur dimérisation ainsi que le recrutement de deux IFNγRII (Greenlung et al., 1993). La formation de ce complexe va permettre la transphosphorylation et l’activation réciproque des JAKs. Les JAKs activées phosphorylent les résidus tyrosine présents sur la partie C-terminale de IFNRI permettant la fixation de STAT1 cytoplasmique via son domaine SH2 (src homology 2) (Greenlung et al., 1994; Heim et al., 1995). La phosphorylation de STAT1 sur son résidu tyrosine Y701 induit la
dissociation rapide du complexe STAT1-récepteur et la formation d’homodimères STAT1 (Shuai et al., 1992, 1993; Greenlung et al., 1995). Cet homodimère est ensuite transloqué vers le noyau où il se fixe sur une séquence consensus appelée l’élément GAS (Gamma activated site) pour induire la transcription de gènes cibles dont IRF-1 (IFN-regulatory factor-1) (Sims et al., 1993, pour revue Schindler et Darnell, 1995). La voie de signalisation JAK/STAT est négativement régulée par plusieurs groupes d’inhibiteurs dont les PIAS (Protein inhibitor of signal transducer and activator of transcription) qui empêchent la liaison des STATs à l’ADN ainsi que les SOCS (Suppressor of cytokine signaling) qui inhibent la phosphorylation des STATs. La synthèse de deux représentants de cette famille, SOCS-1 et 3 est tout particulièrement induite par l’IFNγ, l’IL-6 ou encore la leptine (Song et Shuai, 1998; Bode et al., 1999; Elias et al., 1999).
Au cours de la réaction inflammatoire, les cytokines sont produites par un grand nombre de cellules présentes sur le site de l’infection. Elles agissent localement via l’activation de voies de signalisation spécifiques pour permettre le recrutement des phagocytes et ainsi l’élimination de l’agent pathogène. Par leur action synergique, les cytokines régulent la réaction inflammatoire locale. Outre leur rôle de « coordinateur » de la réaction immunitaire, elles sont capables d’agir dans tout l’organisme grâce à leur propriété endocrine pour réguler les composantes systémique et centrale de la réponse inflammatoire.
Résumé
- Les cytokines coordonnent la réponse inflammatoire afin d’éliminer l’agent pathogène. - Une infection aiguë ou chronique entraîne une production massive de cytokines dont
Introduction
II- Rôles des cytokines dans la réponse systémique et centrale
1- Composante systémique et centrale
La réaction inflammatoire ne reste pas localisée mais retentit sur l’ensemble de l’organisme par l’intermédiaire des cytokines libérées via la circulation sanguine (Figure 1). Les modifications systémiques engendrées se caractérisent par une diminution des concentrations plasmatiques de fer et de zinc, ainsi qu’une augmentation des taux de certaines protéines hépatiques présentes dans le sang et appelées protéines de la phase aiguë (Baumann, 1989). Ces protéines, dont la principale est la protéine C réactive, contribuent à la régulation de l’inflammation (Berczi et al., 2000). Il est également observé une altération des métabolismes lipidique, protéique, glucidique et plus particulièrement l’induction d’une hypertriglycéridémie, une glucogénèse et un catabolisme accru des protéines (Grunfeld et Feingold, 1992; Wilmore, 1974; Lennie et al., 1995; Moldawer et al., 1988).
Les modifications survenant au niveau cérébral ne sont pas le simple reflet de la réaction inflammatoire périphérique, mais correspondent à une réponse spécifique ayant pour objectif de coordonner la composante systémique afin de retourner à l’homéostasie. La fièvre en est une des conséquences les plus évidentes. Elévation de la température corporelle, la fièvre est utile à l’organisme car elle permet d’inhiber la prolifération des microorganismes (Kluger, 1991). La fièvre a été le premier effet attribué à une action centrale des cytokines inflammatoires telles que l’IL-1β, l’IL-6 et le TNFα (Dinarello, 1999).
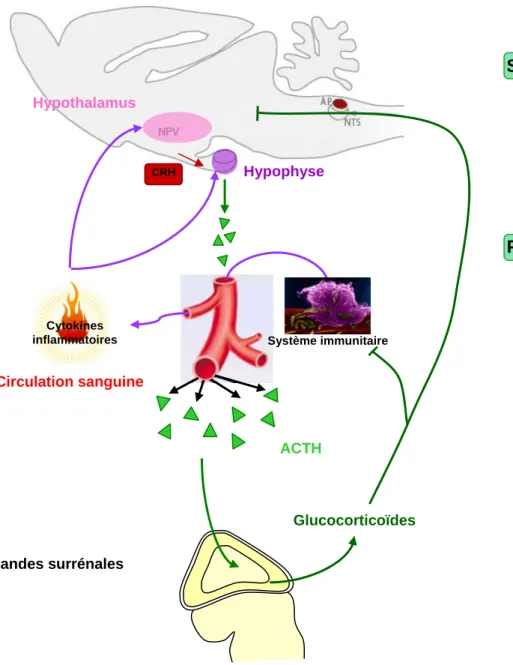
Les cytokines inflammatoires connues pour activer l’axe
hypothalamo-hypophyso-cortico-surrénalien ou axe corticotrope (HPA), induisent la libération de glucocorticoïdes
dans la circulation sanguine (Tilders et al., 1994 ; Turnbull et Rivier, 1999). Stimulé par le stress, l’axe HPA libère la CRH (Corticotrophin-releasing hormone) par l’hypothalamus qui induit à son tour la synthèse d’ACTH (Adrenocorticotrophic hormone) par les cellules de l’hypophyse antérieure. Libérée dans la circulation sanguine, l’ACTH entraîne la synthèse et la libération des glucocorticoïdes par la glande surrénale (Figure 5). L’activation de l’axe HPA par le lipopolysaccharide (LPS), une endotoxine bactérienne activant l’immunité innée, est dépendante de l’IL-1β (Rivier et al., 1989). L’IL-1β stimule l’expression de la CRH et l’IL-1β, l’IL-6 et le TNFα augmentent les taux d’ACTH et de corticostérone plasmatiques (Berkenbosch et al., 1987; Matta et al., 1992; Van der Meer et al., 1996 ; Matta et al., 1993 ; Bernardini et al., 1990). En retour, les glucocorticoïdes inhibent la synthèse des cytokines en agissant directement sur leur cellules productrices (Akira et al., 1990; Baybutt et Holsboer, 1990).
Figure 5: Communication entre les systèmes nerveux,
immunitaire et neuroendocrinien
ACTH: Adrenocorticotrophic hormone, AP: Area postrema, CRH: Corticotrophin-releasing hormone, NPV: Noyau paraventriculaire, NTS: Noyau du tractus solitaire,
SNC: Système nerveux central
Circulation sanguine ACTH CRH Hypophyse Hypothalamus Glandes surrénales Glucocorticoïdes Cytokines inflammatoires SNC Périphérie Système immunitaire
Introduction
Les cytokines ont également des effets sur la neurotransmission monoaminergique et en particulier sur la sérotonine (5-HT), la noradrénaline (NA) et la dopamine (DA). En effet, l’administration d’IL-1β induit une augmentation du turnover des systèmes 5-HT, NA et DA dans l’hypothalamus, l’hippocampe, ainsi que le noyau accumbens (Dunn, 1988; Kabiersch et al., 1988; Linthorst et al., 1995; Lacosta et al., 1994; Song et al., 1999). De plus, ces mêmes effets sont également observés après l’injection de LPS et peuvent être atténués par l’administration d’IL-1Ra (Linthorst et al., 1995; Dunn et al., 2000). Outre l’IL-1β, d’autres cytokines telles que l’IL-6, le TNFα et l’IL-2 sont également capables d’affecter l’activité de la 5-HT, NA et DA dans différentes régions cérébrales dont le cortex préfrontal, l’hypothalamus et l’hippocampe (Zalcman et al., 1994; Pauli et al., 1998).
2- Communication système immunitaire/ système nerveux
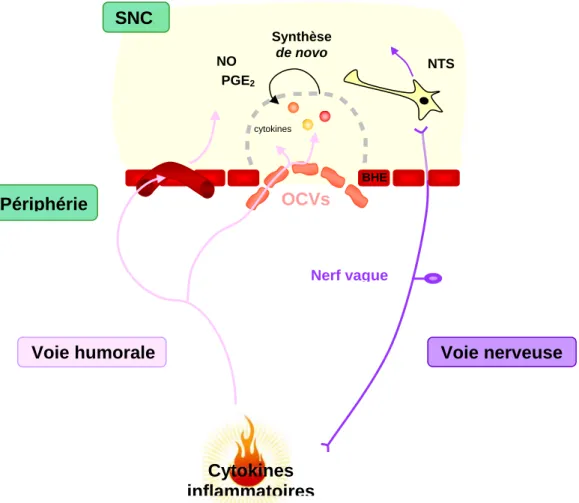
Le cerveau a longtemps été considéré comme un organe privilégié sur le plan immunologique, puisque la barrière hématoencéphalique (BHE) et ses jonctions serrées ne permettent pas le passage des cellules du système immunitaire spécifique. Cependant, le cerveau possède son propre système de défense qui peut rapidement se mettre en état d’alerte à la moindre infection systémique (Figure 6). Il existe certaines régions du cerveau appelées organes circumventriculaires (OCVs) où les vaisseaux sanguins ne possèdent pas de jonctions serrées. Ainsi les cytokines peuvent être passivement transportées du sang vers le parenchyme cérébral. Ce mécanisme de communication entre le cerveau et le système immunitaire constitue la voie humorale (Watkins, 1995). La réaction cérébrale débute au niveau des cellules endothéliales cérébrales, ainsi qu’au niveau des OCVs où les récepteurs au LPS ou l’IL-1β ont été identifiés (Lacroix et al., 1998; Laflamme et Rivest, 2001), puis la réaction s’étend progressivement dans tout le tissu cérébral. L’interaction des cytokines ou du LPS avec leurs récepteurs respectifs conduit en particulier à l’activation des cellules microgliales et astrocytaires cérébrales possédant certaines des propriétés des cellules de l’immunité innée. Comme en périphérie, la réponse immunitairecérébrale est alors médiée par la production de molécules solubles telles que les prostaglandines, les radicaux libres ou encore les cytokines (Brochu et al., 1999; Nadeau et Rivest, 1999, 2000; Quan et al., 1998). Elle régule ainsi l’activité neuronale du cerveau afin d’induire les réponses cérébrales associées à l’inflammation périphérique.
La seconde voie de communication entre les systèmes immunitaire et nerveux dite
voie nerveuse met en jeu l’activation périphérique du nerf vague (Dantzer, 1994). Il a été
Cytokines inflammatoires
Nerf vague
Figure 6: Les voies de communication entre le système
immunitaire et le système nerveux central
BHE: Barrière hémato-encéphalique, NO: monoxyde d’azote, NTS: Noyau du tractus solitaire, OCVs: Organes circumventriculaires, PGE2: Prostaglandines de type E2, SNC: système nerveux central
OCVs BHE cytokines Synthèse de novo PGE2 NO SNC NTS Périphérie
Introduction
démontré que les neurones sensoriels du nerf vague expriment les récepteurs de l’IL-1 et leur activité électrique peut être stimulée par l’IL-1β circulante via la synthèse de prostaglandines (Ek et al., 1998; Niijima, 1996). Les afférences vagales, en projetant vers le noyau du tractus solitaire (NTS) ainsi que vers l’hypothalamus, pourraient sous-tendre la production cérébrale d’IL-1 au niveau du NTS et l’activation des neurones NA et DA de l’hypothalamus (Gordon, 2000; Elmquist et Saper, 1996). Des animaux vagotomisés présentent une atténuation des modifications comportementales normalement observées après injection de LPS confirmant ainsi l’implication du nerf vague dans la réponse centrale à l’inflammation (Bluthé et al., 1994).
3- Conséquences de l’action régulée des cytokines sur le cerveau
Les cytokines sont responsables de l’apparition des symptômes non spécifiques de la maladie incluant la fièvre ainsi que des changements physiques et comportementaux profonds tels qu’une grande fatigue, une apathie, une difficulté à se concentrer, une perte d’appétit, des troubles du sommeil, une réduction de l’intérêt pour l’environnement physique et social, une anhédonie (Hart, 1988; Dantzer et al., 1998). L’ensemble de ces modifications regroupées sous le terme de comportement de maladie n’est pas le reflet de processus délétères, mais représente un état motivationnel (Hart, 1988; Kluger, 1991; Dantzer et al., 2006). En effet, il s’agit d’une réorganisation des priorités de l’organisme afin de lutter efficacement contre l’agent pathogène responsable de l’infection.
L’administration systémique et centrale de cytokines recombinantes ou de LPS permet de reproduire l’ensemble des symptômes non spécifiques de la maladie (Kent et al., 1992; Larson et Dunn, 2001; Dantzer, 2001). Chez le rat, l’injection systémique d’IL-1β ou de TNFα induit l’apparition d’un état fébrile, une réduction du temps d’exploration sociale ainsi qu’une diminution de la prise alimentaire (Bluthé et al., 2000). De plus, les patients soumis à une immunothérapie par injections d’IL-2 ou d’IFNα dans le cadre de traitements anticancéreux, ou antiviraux dans le cas de l’hépatite C, développent rapidement des symptômes neurovégétatifs et somatiques semblables aux symptômes de la grippe: fatigue, anorexie, troubles du sommeil et de l’appétit, douleur, troubles gastro-intestinaux (Bonaccorso et al., 2001; Capuron et al., 2000). Si ces manifestations sont majoritairement transitoires et disparaissent dès l’arrêt du traitement, l’administration prolongée de cytokines provoque, dans un tiers des cas, l’apparition de symptômes neuropsychiatriques comprenant des troubles de l’humeur (état dépressif, anhédonie, irritabilité, anxiété), ainsi que des
troubles cognitifs (déficit d’attention, perte de mémoire, confusion, indécision) (Bonaccorso et al., 2002; Capuron et al., 2002a). Chez l’homme, il a été montré que l’administration de LPS à des volontaires sains induit une augmentation marquée des taux circulants de cytokines et de glucocorticoïdes positivement corrélée au niveau d’anxiété, de dépression, ainsi que de déficits cognitifs (Yirmiya et al., 2000). Parmi les troubles de l'humeur (dépression et anxiété) observés chez les patients soumis à un traitement par l’IFNα, les symptômes dépressifs sont les plus couramment décrits (60%). Les troubles cognitifs ne sont quant à eux, rapportés que chez 30% des patients (Capuron et al., 2002a). Ainsi, ces résultats suggèrent que l’action chronique des cytokines pourrait être responsable du développement des symptômes dépressifs observés lors d’une infection.
Résumé
- En plus de leur action locale, les cytokines sont capables grâce à leur propriété endocrine
d’agir sur le SNC pour réguler la composante centrale de la réaction inflammatoire.
- Les cytokines agissent en central par l’intermédiaire des voies humorale et nerveuse et
entraînent entre autre, une hyperactivité de l’axe HPA, une altération de la neurotransmission monoaminergique, en particulier 5-HT, ainsi que des changements comportementaux regroupés sous le terme de comportement de maladie.
- Ces altérations comportementales sont semblables aux symptômes neurovégétatifs induits
par l’administration de cytokines chez l’homme et parfois associés à l’apparition de troubles de l’humeur.
♦♦♦
Développement des troubles de l’humeur : conséquences centrales
de l’action dérégulée des cytokines
I- Description de la symptomatologie de la dépression
Le terme dépression du latin depressio, enfoncement, désigne une maladie dont la manifestation centrale est un état mental défini par une lassitude importante, une dépréciation de soi ou encore des pensées pessimistes. La dépression est décrite comme une association de symptômes psychiques et somatiques (Lôo et Lôo, 2001). Elle représente la forme la plus
Introduction
courante des désordres psychiatriques et affecte 20% de la population, indépendamment de l’âge, du statut socio-économique ou du style de vie. Le diagnostic est une étape importante de la prise en charge des maladies dépressives et nécessite d’identifier les nombreux symptômes, ainsi que de préciser leur évolution. Un système de critères diagnostiques exclusivement descriptif a donc été mis en place et défini par le DSM (Diagnostic and Statistical Manual of Mental Disorders, 1952). Ainsi, le diagnostic d’une dépression majeure est effectif quand cinq des neuf symptômes définis par le DSM IV sont présents presque quotidiennement, pendant au moins 15 jours et dont au moins un des symptômes est soit une humeur dépressive, soit une perte d’intérêt ou de plaisir (DSM Fourth Edition, 1994) (Figure7).
II- Etiologie de la dépression
1- Théorie neuroendocrinienne, neurochimique et anatomique
Outre les caractéristiques psychologiques, de nombreuses études ont établi une composante neurobiologique de la dépression. Ainsi, il a pu être observé une hyperactivité de l’axe HPA chez les patients dépressifs (Ströhle et Holsboer, 2003). En effet, ces patients présentent une augmentation des taux plasmatiques et urinaires de cortisol, ainsi qu’une élévation marquée des concentrations de CRH dans le liquide céphalo-rachidien (Maes et al., 1998; Banki et al., 1987). Le cortisol étant incapable de freiner la production d’ACTH et de CRH, l’hypothèse d’un dysfonctionnement du rétrocontrôle négatif de l’axe HPA dans la dépression a été suggérée (Sheline, 2000; Pariante, 2003). De plus, cette altération serait due à une réduction de la fonction des récepteurs aux glucocorticoïdes (Modell et al., 1997; Pariante, 2004).
L’implication des monoamines dans la dépression a été proposée après l’observation que certaines substances capables d’induire leur déplétion entraînaient le développement de symptômes dépressifs (Lemieux et al., 1956). De plus, la dépression serait associée à des altérations de la production cérébrale de 5-HT, NA et DA (Schildkraut et al., 1965; Coppen, 1967; Lambert et al., 2000). Une réduction des concentrations en NA et 5-HT a été observée dans le liquide céphalo-rachidien de patients dépressifs (Bourne et al., 1968; Beskow et al., 1976). De plus, des études post-mortem ont montré des variations de l’expression des différents sous-types de récepteurs sérotoninergiques et en particulier une augmentation de la densité des récepteurs 5-HT1A (Arora et Meltzer, 1989; Arango et al., 1995; Mann, 1999).
Enfin, le fait que les traitements pharmacologiques affectant le système sérotoninergique
Humeur dépressive pendant la majeure partie de la journée
Anhédonie: diminution très nette de l’intérêt ou du plaisir lors
d’activités normalement plaisantes
Perte ou gain de poids significatif
Troubles du sommeil (insomnie ou hypersomnie)
Agitation ou ralentissement psychomoteur
Fatigue ou perte d’énergie
Sentiments de dévalorisation, faible estime de soi ou culpabilité excessive
Diminution de la capacité de réflexion et de concentration
Pensées pessimistes, de mort et de suicide récurrentes
⇒
Au moins cinq des symptômes doivent être présents presque quotidiennement pendant au moins deux semaines consécutives⇒
Au moins un des symptômes doit être soit une humeur dépressive, soit une perte d’intérêt ou de plaisir (anhédonie)Introduction
atténuent les symptômes dépressifs a permis de focaliser les recherches sur l’implication de la 5-HT (Blier, 2003).
La dépression est également associée à des modifications morphologiques et fonctionnelles cérébrales. Des études d’imagerie cérébrale réalisées chez des patients dépressifs ont permis de mettre en évidence des altérations du flux sanguin cérébral, ainsi que du métabolisme du glucose dans certaines régions du cerveau telles que le cortex préfrontal et cingulaire, l’amygdale, le striatum, le thalamus ou encore l’hippocampe (Drevets, 2000a, 2000b, 2001; Liotti et Mayberg, 2001). De plus, une réduction du volume du cortex préfrontal et de l’hippocampe a été observée lors d’études post-mortem et corrélée à une atrophie cellulaire et/ou une perte neuronale et gliale (Rajkowska, 2000; Drevets, 2001 ; Manji et Duman, 2001). Chez l’animal, l’ensemble de ces modifications comportementales, neuroendocriniennes et neurochimiques a été reproduit dans des modèles de dépression tel que la lésion du bulbe olfactif (Jesberger et Richardson, 1986; Kelly et al., 1997). Ces animaux présentent notamment une diminution des épines dendritiques des régions CA1, CA3 et du gyrus denté de l’hippocampe (Norrholm et Ouimet, 2001), ainsi qu’une perturbation de la réponse immunitaire et plus particulièrement une réduction de la prolifération des lymphocytes (Song et Leonard, 2005).
2- Théorie immunitaire
La relation entre le système immunitaire et la dépression a fait l’objet d’un certain nombre de travaux démontrant que la dépression était associée à une altération du fonctionnement du système immunitaire (Asnis et Miller, 1989; Kronfol et al., 1983; Schleifer et al., 1984). In vitro, il a été montré que cette immunosuppression se caractérisait par une réduction de la prolifération des lymphocytes T et B ainsi que celle de l’activité des cellules NK (Schleifer et al., 1984; Irwin et Gillin, 1987). De plus, il a pu être observé une production plasmatique accrue de cytokines inflammatoires: l’IL-1β, l’IL-2, l’IL-6, l’IL-10 et l’IFNγ (Seidel et al., 1995, 1999), ainsi qu’une augmentation des concentrations d’IL-1β et d’IL-6 dans le liquide céphalo-rachidien de patients souffrant de troubles dépressifs (Levine et al., 1999). Cette forte production de cytokines étant corrélée à la sévérité des troubles de l’humeur, il a été proposé que l’activation du système immunitaire pourrait être impliquée dans la pathogenèse de la dépression majeure (Maes, 1995, 1999). De plus, l’observation qu’un tiers des patients atteints de cancers ou encore d’une hépatite C et soumis à une immunothérapie développent des troubles de l’humeur, étaye la théorie d’une relation entre
cytokines et dépression (Bonaccorso et al., 2002; Capuron et al., 2002b). L’intensité de la dépression induite par l’injection intraveineuse d’IL-2 ou d’IFNα est corrélée positivement avec la diminution des concentrations plasmatiques de tryptophane, l’acide aminé précurseur de la 5-HT. D’autre part, la paroxetine, inhibiteur sélectif de la recapture de la 5-HT administrée en prévention avant le début de l’immunothérapie prévient le développement des symptômes dépressifs (Capuron et al., 2002b).
Des altérations de l’humeur ont pu être observées dans un certain nombre de pathologies comprenant une composante inflammatoire dont l’arthrite rhumatoïde, l’athérosclérose, les cancers, la maladie coronarienne. En particulier, les patients atteints du virus de l’immunodéficience humaine (VIH) présentent un syndrome inflammatoire chronique ainsi que des atteintes neurologiques accompagnées de troubles psychiatriques dont le syndrome anxiodépressif (Glass et Jonhson, 1996; Dilley et al., 1986).
Résumé
- La dépression est une pathologie psychiatrique multifactorielle associant symptômes
psychiques et somatiques.
- A ces symptômes s’ajoutent des modifications neurobiologiques telles qu’une hyperactivité
de l’axe HPA, une dérégulation du système 5-HT, une altération de la plasticité cérébrale ainsi qu’une production plasmatique accrue de cytokines inflammatoires.
- La présence de cytokines dans le cadre d’une immunothérapie ou d’une pathologie à
composante inflammatoire serait un facteur favorisant le développement de troubles de l’humeur.
Introduction
Cytokines et altération du métabolisme du tryptophane :
implication potentielle dans le développement des troubles de
l’humeur
I- Rôle du tryptophane dans le métabolisme de la sérotonine
1- Rôle et distribution de la sérotonine
La 5-Hydroxytryptamine ou 5-HT initialement localisée au sein des plaquettes et connue pour son activité vasoconstrictrice, a été isolée et purifiée pour la toute première fois en 1948 à partir du sérum (Rapport et al., 1948). Bien que 95% de la 5-HT de l’organisme soient produits par les cellules entérochromaffines de la muqueuse du tractus gastro-intestinal, elle a également été mise en évidence au niveau cérébral (Erspamer et Asero, 1952; Twarog et Page, 1953). Les études menées par le groupe d’Amin et al., en 1954 ont montré une distribution différentielle de la 5-HT au sein du système nerveux central et exclusivement au sein de la substance grise contenant les corps cellulaires des neurones. Il a ensuite été démontré par immunohistochimie que la 5-HT était synthétisée par des neurones dont les corps cellulaires sont particulièrement concentrés dans les noyaux du raphé au sein du tronc cérébral (Dahlströem et Fuxe, 1964 ; Steinbusch, 1981 ; Touret et al, 1987). Les neurones sérotoninergiques les plus rostraux projettent majoritairement dans le cerveau antérieur, le thalamus ainsi que l’hypothalamus par des voies ascendantes, tandis que les cellules les plus caudales et ventrales projettent par des voies descendantes dans la mœlle épinière et le cervelet respectivement (Azmitia, 2007). Ainsi, la 5-HT est largement distribuée au sein du système nerveux central où elle joue le rôle de neurotransmetteur. Elle est impliquée dans la régulation de la plupart des grandes fonctions de l’organisme telles que la régulation du cycle veille-sommeil, la thermorégulation, les comportements alimentaires (effet anorexigène) ainsi que sexuels, les états émotionnels tels que l’humeur (action anti-dépressive) et l’anxiété, la nociception et les contrôles moteurs (pour revue Lucki, 1998).
2- Biosynthèse et métabolisme
La 5-HT ne pouvant traverser la BHE, sa synthèse au niveau cérébral est exclusivement réalisée au sein des neurones sérotoninergiques (Udenfriend et al., 1957). Le
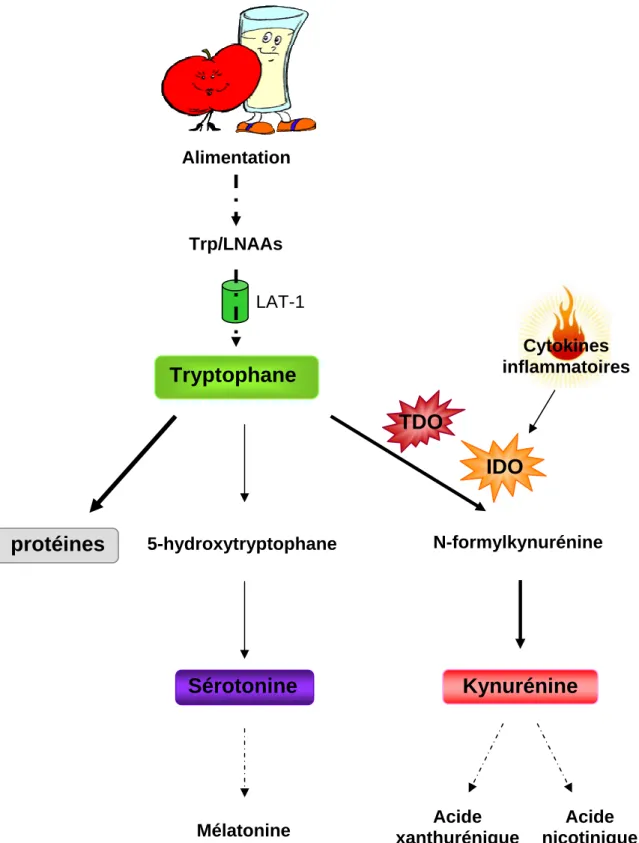
tryptophane (Trp), précurseur de la biosynthèse de la 5-HT, est un acide aminé dit essentiel,
c'est-à-dire qu’il ne peut être synthétisé par l’organisme, mais doit exclusivement être apporté via la dégradation des protéines provenant de l’alimentation (Figure 8). La quantité quotidienne de Trp ingérée varie de 0.5 à 1 g, bien que l’apport minimum nécessaire soit
Figure 8: Les différentes voies de catabolisme du tryptophane
IDO: Indoléamine 2,3-dioxygénase, LAT-1: L-type amino acid transporter 1, LNAAs: Large neutral amino acids, TDO: Tryptophane 2,3-dioxygénase, Trp: Tryptophane
Alimentation Trp/LNAAs LAT-1
Tryptophane
protéines
5-hydroxytryptophaneSérotonine
Cytokines inflammatoiresTDO
IDO
N-formylkynurénineKynurénine
Acide AcideIntroduction
évalué à 200 mg/j. Plus de 95% du Trp est utilisé pour la synthèse des protéines, cependant il ne représente que seulement 1% des 20 acides aminés constituant les protéines. Après dégradation des protéines de l’alimentation, le Trp est présent dans la circulation sanguine sous deux formes : la majeure partie (50 à 80%) est liée à l’albumine sérique (McMenamy et Oncley, 1958), tandis que le reste du Trp reste sous forme libre, 1% étant utilisé pour la synthèse de 5-HT (Salter et al., 1989). En effet, seule la fraction libre du Trp peut traverser la BHE grâce à un transporteur actif, le L-type amino acid transporter 1 (LAT-1) présent sur la membrane de certaines populations de cellules, principalement les cellules endothéliales (Curzon, 1996 ; pour revue, Russo et al., 2003).
La biosynthèse de la 5-HT à partir de son précurseur est réalisée en 2 étapes distinctes (Boadle-Biber, 1993): une hydroxylation du Trp en 5-hydroxytryptophane (5-HTP), réaction catalysée par l’enzyme limitante de cette voie de biosynthèse, la tryptophane-5-hydroxylase (Grahame-Smith, 1964, Lovenberg et al., 1967), suivie d’une décarboxylation du HTP en 5-HT par la L-amino-décarboxylase (Henry et Bowsher, 1986). Cependant, la voie de biosynthèse ne se termine pas avec la formation de HT car une faible concentration de la 5-HT produite est elle-même transformée en mélatonine au sein de la glande pinéale (Moore et Rapport, 1971; Bowsher et Henry, 1983). Après dépolarisation du neurone, la 5-HT libérée dans la fente synaptique est rapidement inactivée en étant recapturée dans le bouton présynaptique via un transporteur membranaire spécifique (SERT) (Rudnick, 1977). Sa dégradation est assurée par la monoamine oxydase de type A (MAO A) présente au sein des neurones, sur la membrane externe des mitochondries (Cawthon et Breakefield, 1979) pour former l’acide 5-hydroxyindole acétique (5-HIAA), principalement retrouvé dans les urines, mais également au niveau du liquide céphalo-rachidien (5%).
3- Modulation du métabolisme de la sérotonine
La biodisponibilité cérébrale du Trp représente le premier facteur limitant de la biosynthèse de la 5-HT. En effet, de nombreux facteurs modulant l’apport du Trp au cerveau peuvent altérer les taux cérébraux de Trp et de ce fait la synthèse de 5-HT (Schiepers et al., 2005).
Lors du passage de la BHE, le Trp entre en compétition avec les autres acides aminés neutres plasmatiques: phénylalanine, tyrosine, valine, leucine et isoleucine (LNAAs) pour accéder au site de liaison du transporteur LAT-1 (Pardridge, 1983; Kanai et al., 1998). Ainsi, l’administration en excès d’un des LNAAs entraîne une diminution du taux de transport du
Trp dans le cerveau (Blasberg et Lajtha, 1966; Murray, 1973; Drewes et al., 1977). De plus, les concentrations des LNAAs sont également sous la dépendance de certaines hormones. En effet, le cortisol réduit les taux plasmatiques libres de Trp et de tyrosine provoquant une diminution du Trp cérébral (Maes et al., 1990). A l’opposé, il a été montré que l’insuline induisait une déplétion des LNAAs autre que le Trp (Wurtman et al,. 1980).
Comme l’enzyme tryptophane 5-hydroxylase n’est pas saturée vis à vis du Trp en conditions physiologiques, les variations plasmatiques du Trp ont un impact direct sur les taux de 5-HT cérébraux (Eccleston et al., 1965; Friedman, 1972). Ainsi, l’administration i.p. aigue ou chronique de Trp provoque une augmentation de la synthèse cérébrale de 5-HT chez le rat et plus particulièrement au niveau de l’hypothalamus (Ashcroft et al., 1965; Knott et Curzon, 1974). A l’inverse, la déplétion en Trp entraîne une réduction des taux sériques et cérébraux de Trp ainsi que de 5-HT et de son catabolite le 5-HIAA, présent dans le liquide céphalo-rachidien chez l’homme et le rat (Young et al., 1989; Biggio al., 1974). Ainsi, toutes ces données supportent l’idée que l’apport cérébral en Trp est un paramètre essentiel à la synthèse de la 5-HT.
Le Trp utilisé pour la synthèse de 5-HT cérébrale ne représente qu’une petite partie du pool de Trp provenant de l’alimentation (Udenfriend et al., 1956). En effet, la majeure partie du Trp n’étant pas utilisée pour la synthèse protéique est catabolisée via la voie de la kynurénine. La première étape de cette voie de dégradation est catalysée par deux enzymes, la
tryptophane 2,3-dioxygénase (TDO) et l’indoléamine 2,3-dioxygénase (IDO) pouvant être
modulées par différents facteurs.
Résumé
- La 5-HT est un neurotransmetteur ubiquitaire, synthétisé à partir du Trp, acide aminé
essentiel provenant uniquement de la dégradation des protéines de l’alimentation.
- La biodisponibilité cérébrale du Trp est le premier facteur limitant de la synthèse de 5-HT.
Ainsi, une diminution des taux de Trp ayant pour conséquence une réduction de la synthèse de 5-HT pourrait indirectement être impliquée dans la mise en place des troubles de l’humeur.