© Bachar Cheaib, 2020
Étude de l’évolution contemporaine de systèmes
microbiens environnementaux et hôtes associés dans
un contexte d’écotoxicologie
Thèse
Bachar Cheaib
Doctorat en biologie
Philosophiæ doctor (Ph. D.)
Québec, Canada
Étude de l’évolution
contemporaine de systèmes
microbiens environnementaux et
hôtes associés dans un contexte
d’écotoxicologie
Thèse
Bachar Cheaib
Doctorat en biologie
Philosophiae doctor (Ph.D.)
Sous la direction de :
ii
Résumé
Les microbes ou micro-organismes sont les producteurs primaires des services écosystémiques pour les cycles biogéochimiques de la terre et les systèmes biologiques. Les xénobiotiques marquent une nouvelle ère anthropogénique « l’anthropocène », et ils représentent une source de sélection artificielle de la structure et de la composition de la biodiversité microbienne. Par conséquent, les perturbations anthropogéniques sont néfastes pour les systèmes microbiens et induisent des changements adaptatifs ou des dommages dans leurs répertoires génotypiques. L’assemblage des communautés microbiennes durant la résistance et la résilience est gouverné par des processus éco-évolutifs.
Ce travail découle de l’intersection transdisciplinaire de l’écotoxicologie, l’écologie microbienne, la métagénomique et la bioinformatique. L’objectif de ce travail consiste à étudier les signatures adaptatives de la résistance et de la résilience microbienne selon deux modèles. Le premier est environnemental (E) composé d’un bassin versant lacustre contaminé par des métaux lourds. Le deuxième modèle est hôte-associé (HA), constitué d’un système expérimental d’exposition de la Perchaude (Perca flavescens) au chlorure de cadmium selon deux régimes constant et graduel.
Trois nouveautés résument les travaux de cette thèse de doctorat. Premièrement, le phénomène de découplage taxon-fonction a été démontré pour la première fois, dans le système E sous un gradient sélectif de pollution, et au sein du microbiote cutané dans le système HA durant sa période de résilience.
Deuxièmement, des altérations significatives de la diversité taxonomiques et fonctionnelles mettent en évidence des signatures adaptatives du résistome et de l’érosion des fonctions métaboliques dans le système E. Quant au système HA, le stress métallique a augmenté la prévalence significative de souches pathogènes et des opportunistes avec une dysbiose cutanée de la perchaude accompagnée par une réduction de sa capacité de résistance à une colonisation bactérienne massive.
iii
Troisièmement, la modélisation de l’assemblage bactérien de microbiote du système HA montre des rôles confondus de l’ontogenèse et de la force de sélection durant la période de résistance. La persistance des effets à long terme de la sélection durant le stade de résilience a été expliquée par une augmentation inattendue de la bioaccumulation du cadmium dans les tissus hépatiques de l’hôte.
En conclusion, nos travaux montrent que l’adaptation des répertoires métagénomiques peut être décelée par le phénomène de redondance fonctionnelle observée à l’échelle de découplage taxon-fonction, ce qui reflète potentiellement une stratégie adaptative par transfert horizontal de gènes partagés entre les communautés microbiennes environnementales sous perturbation graduelle.
Dans le système HA, l’assemblage de microbiote montre un gradient de processus neutres et non neutres. Enfin, la dérive taxonomique serait une force écologique non négligeable plus importante dans le système environnemental que dans le système intestinal durant et après la perturbation.
iv
Abstract
Microbes or microorganisms are the primary producers of ecosystem services for biogeochemical cycles of the earth and biological systems. Xenobiotics mark a new anthropogenic era, "the Anthropocene," and they represent a source of artificial selection of the structure and composition of microbial biodiversity. As a result, anthropogenic disturbances are detrimental to microbial systems and induce adaptive changes or damage in their metagenomic repertories. During resistance and recovery, the ecological processes governing the assembly of microbial communities cannot be dissociated from those of microbial evolution.
This work stems from the transdisciplinary intersection of ecotoxicology, microbial ecology, metagenomics and bioinformatics. The main goal is to understand the adaptive signatures of microbial resistance and resilience in two models. The first is environmental (E) composed of a lake-bound watershed contaminated by heavy metals. The second model is host-associated (HA), consisting of an experimental system of perch (Perca
flavescens) intoxicated with cadmium using two steady and gradual regimes.
Three novelties summarize the work of this doctoral thesis. Firstly, the phenomenon of taxon-function decoupling has been demonstrated for the first time, in the E system under selective pollution gradient, and second, within the cutaneous microbiota in the HA system during its recovery stage. Third, the microbiota assembly modelling in the HA system suggested mixed effects of ontogenesis, and selective pressure during the period of resistance and recovery. The increase in cadmium bioaccumulation in liver tissues of perch can argue the persistence of the long-term effects of selection during the recovery stage.
In conclusion, our work showed that the adaptation of microbial metagenomic repertories could be revealed through functional and taxonomic redundancy patterns observed at the scale of taxon-function decoupling. The gap between functional and taxonomic diversity reflects an adaptive strategy by horizontal gene transfer among environmental communities microbial under gradual disruption.
v
In the HA system, the microbiota assembly shows a gradient of neutral and non-neutral processes. Finally, the taxonomic drift is a significant ecological force, more effective in the environmental system than in the intestinal system during and after the disruption.
vi
Table des matières
Résumé ... ii
Abstract ... iv
Liste des tableaux ... x
Liste des Schémas ... xi
Liste des figures ... xii
Liste des abréviations ... xiii
Remerciements ... xvi
Introduction Générale ... 1
L’anthropocène: des effets anthropiques sur la biodiversité et l’assemblage des communautés microbiennes ... 2
Les questions et les chapitres de cette thèse ... 4
Chapitre 1 – Méthodes et concepts de base en écologie
microbienne ... 8
1.1 Les microbes sont ubiquitaires et l’environnement les filtre ... 9
1.2 Accès à la biodiversité microbienne, de la boite de Pétri au métagénome ... 9
1.3 L’avènement du séquençage de nouvelle génération ... 10
1.4 Les approches de la métagénomique ... 12
1.5 L’approche d’amplicons basée sur un gène marqueur universel ... 12
1.6 L’approche métagénomique globale basée sur le séquençage de l’ADN total. ... 14
1.7 Approche de l’opération taxonomique universelle (UTO) ... 14
1.8 L’approche des variants des séquences d’amplicons (VSA).... 16
1.9 Les mesures écologiques et phylogénétiques de la diversité. 17 1.9.1 Les mesures de la diversité alpha ... 17
1.9.2 Les mesures de la diversité Beta ... 19
1.9.2.1 Les distances écologiques ... 19
1.9.2.2 Les distances phylogénétiques ... 20
1.10 Réseaux d’interactions microbiennes ... 20
1.11 Les quatre forces évolutives et processus écologiques ... 22
1.11.1 La mutation ... 22
1.11.2 La sélection ... 24
1.11.3 La migration ... 26
1.11.4 La dérive génétique ... 27
1.11.5 Interactions des forces évolutives ... 28
1.11.6 De l’évolution à l’écologie ... 30
1.11.6.1 L’adaptation Locale ... 30
1.11.6.2 L’adaptation rapide ... 31
vii
1.13 Introduction à la modélisation mathématique en écologie et
évolution microbienne... 33
1.13.1 Préambule historique de la modélisation ... 34
1.13.2 Aperçu sur les modèles d’assemblage microbiens ... 37
1.13.2.1 Les modèles métaboliques ... 37
1.13.3 Les modèles kinétiques ... 38
1.13.4 Les modèles spatiaux ... 38
1.13.4.1 Modèles basés à l’échelle individuelle ... 39
1.13.4.2 Modèles basés à l’échelle populationnelle ... 39
1.13.4.3 Modèles basés à l’échelle de communauté ... 40
1.13.5 Les modèles neutres en écologie microbienne ... 40
1.13.6 Le modèle neutre de Sloan ... 42
1.13.7 Neutralisme et déterminisme dans l’assemblage du microbiote de l’hôte. ... 45
1.13.8 Les modèles éco-évolutifs ... 46
1.14 Récapitulatif ... 48
Chapitre 2: Taxon-function decoupling as an adaptive
signature of lake microbial metacommunities under a chronic
polymetallic pollution gradient ... 50
2.1 Résumé ... 51
2.2 Abstract ... 52
2.3 Introduction ... 53
2.4 MATERIALS AND METHODS ... 57
2.4.1 Lake characteristics and locations ... 57
2.4.2 Metallic and chemical gradient surveys ... 58
2.4.3 Water sampling ... 58
2.4.4 DNA extraction and metagenome sequencing ... 58
2.4.5 Bioinformatic and statistical analysis ... 58
2.5 Results ... 61
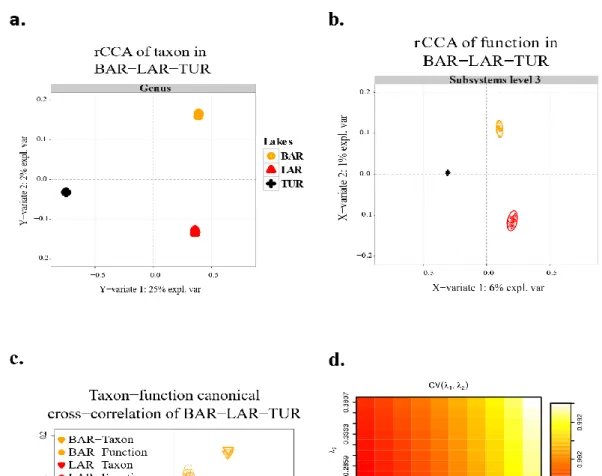
2.5.1 Decoupling taxon-function ... 61
2.5.2 Detangled taxonomic structure and function diversity ... 62
2.5.3 Canonical correlations of taxon and function ... 62
2.5.4 Taxonomic variation signatures... 63
2.5.5 Role of trace metals in taxonomic variation signatures ... 64
2.5.6 Function variation signatures ... 64
2.5.7 Role of trace metals in function variation signatures ... 66
2.6 Discussion ... 67
2.6.1 Decoupling taxon-function as a signature of adaptive strategies ... 67
2.6.2 Taxonomic adaptive signatures ... 69
2.6.3 Functional adaptive signatures ... 71
2.7 Conclusions ... 73
2.8 Figures ... 74
2.9 Supplementary figures ... 82
2.10 Supplementary Material ... 94
Chapitre 3: From networks to models: The Yellow Perch
(Perca flavescens) microbiome assembly under metal toxicity
... 96
viii
3.2 Abstract ... 98
3.3 Introduction ... 99
3.4 Materials and methods ... 102
3.4.1 Fish rearing. ... 102
3.4.2 Exposure regimes to cadmium. ... 102
3.4.3 Host-microbiota and water sampling. ... 102
3.4.4 Metal concentration in water and fish liver. ... 103
3.4.5 DNA extraction to Illumina Miseq sequencing. ... 103
3.4.6 Analysis of 16S rDNA amplicons. ... 104
3.4.7 Correlational networks. ... 104
3.4.8 Metacommunity assembly modelling. ... 105
3.5 Results ... 105
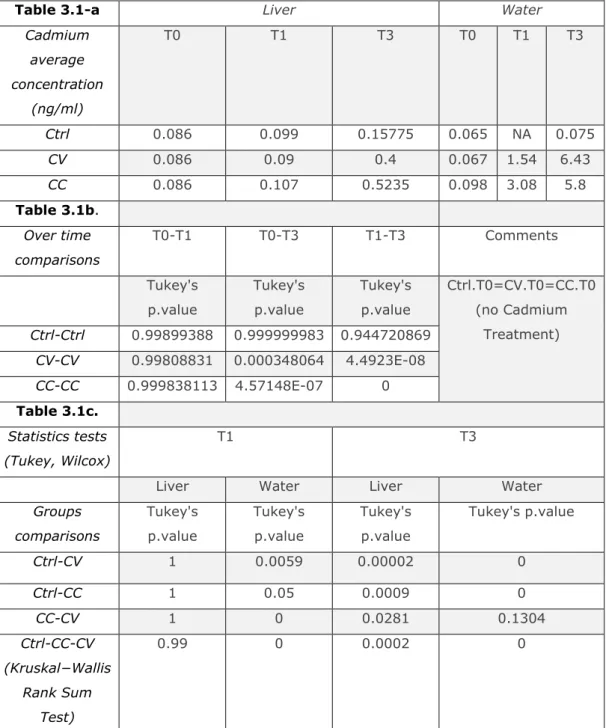
3.5.1 Metal concentrations in water and host livers ... 105
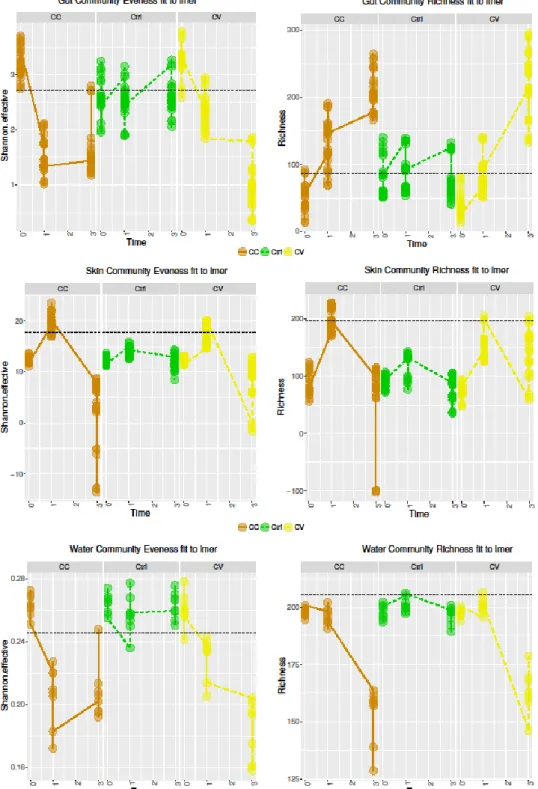
3.5.2 Mixed Effects of time and treatment on metacommunity alpha diversity ... 106
3.5.3 An important effect of time on the taxonomic composition of metacommunities ... 106
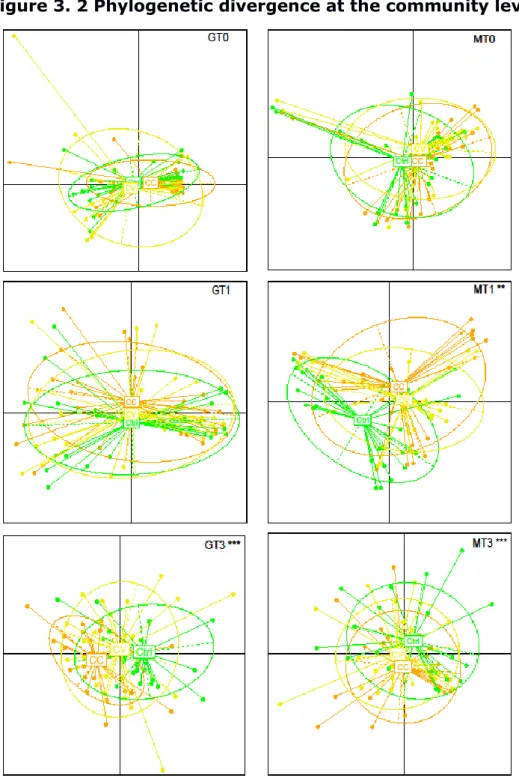
3.5.4 Community-level phylogenetic divergence ... 107
3.5.5 Correlational metacommunity networks ... 107
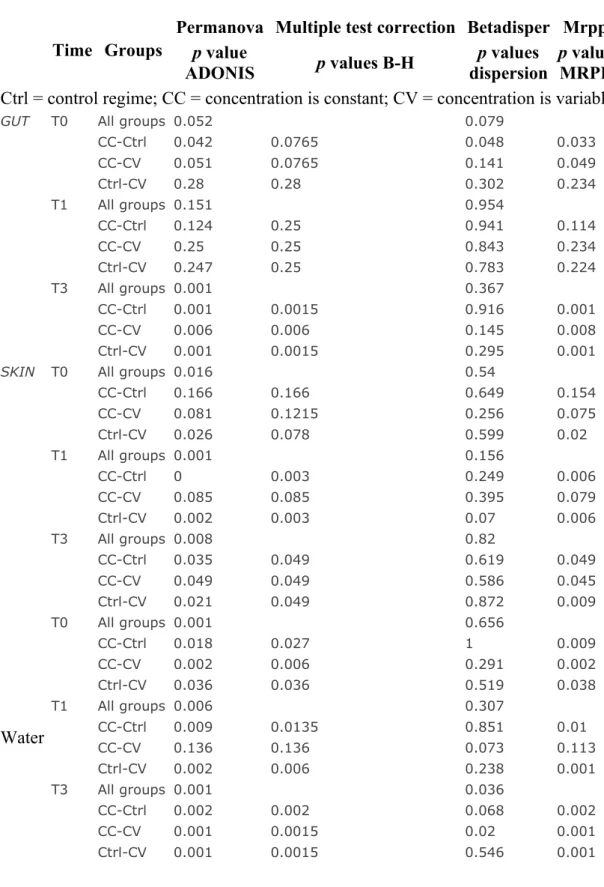
3.5.5.1 Substantial role of rare taxa in the metacommunity network connectivity ... 107
3.5.5.2 Reduced network connectivity in gut communities under cadmium stress ... 107
3.5.5.3 Negative correlations in Skin Mucous Community networks suggest dysbiosis ... 108
3.5.5.4 Fragmentation of water microbial community networks . 108 3.5.6 Stochasticity in water community assembly and determinism in that of host microbiota ... 109
3.6 Discussion ... 109
3.6.1 Phylogenetic divergence at the community-level revealed the impact of Cadmium exposure. ... 110
3.6.2 Gradual disconnection of abundant taxa from the main gut interacting network. ... 111
3.6.3 Rare OTUs play a pivotal role in community assembly. ... 112
3.7 Conclusions ... 112 3.8 Perspective ... 113 3.9 Tables ... 114 3.10 Figures ... 119 3.11 Supplementary Figures ... 125 3.12 Supplementary Material ... 131
Chapitre 4: Community recovery dynamics in yellow perch
microbiome after gradual and constant metallic perturbations
... 134
4.1 Resumé ... 135 4.2 Abstract ... 136 4.3 Introduction ... 137 4.4 Methods ... 139 4.4.1 Fish rearing ... 1394.4.2 Exposure regimes to cadmium ... 140
ix
4.4.4 Host-microbiota and water sampling ... 140
4.4.5 Metal concentration in water and fish liver ... 141
4.4.6 DNA extraction, libraries preparation and 16S amplicons sequencing ... 141
4.4.7 Bioinformatics and biostatistics analyses ... 141
4.4.7.1 Reads preprocessing and OTUs clustering ... 141
4.4.7.2 Post-OTUs analysis, networks and function prediction. ... 143
4.5 Neutral and deterministic models to asses the recovery of community assembly. ... 144
4.6 Results ... 144
4.6.1 Cadmium concentration bioaccumulation in the fish liver during recovery time ... 144
4.6.2 Genotypic signatures of community recovery ... 145
4.6.3 Microbial taxonomic composition change during recovery 146 4.6.4 Correlational networks of host and water microbiome ... 148
4.6.5 Recovery of microbial functional diversity a time T5. ... 149
4.6.6 The role of neutral and deterministic processes in the recovery of host microbiota ... 149
4.7 Discussion ... 150 4.8 Conclusions ... 154 4.9 Tables ... 155 4.10 Figures ... 158 4.11 Supplementary figures ... 166 4.12 Supplementary Material ... 168
Discussion et conclusions générales... 170
x
Liste des tableaux
Table 3. 1 Statistics’ summary of Cadmium concentration variation over time and treatments in water tanks and fish livers. ... 114 Table 3. 2 Statistical summary of alpha-diversity changes over time and treatments. ... 115 Table 3. 3 Phylogenetic divergence at the community level. ... 117
Table 4. 1 Statistics of Cd concentrations in water and fish liver over time and treatments. ... 155 Table 4. 2 Phylogenetic divergence in host and water microbiomes. ... 156
xi
Liste des Schémas
Schéma 1. 1 Approches métagenomiques de l’analyse de microbiote. ... 15 Schéma 1. 2. Boite noire de l’écologie des communautés. ... 33 Schéma 1. 3. Espace des modèles éco-évolutifs. ... 48
xii
Liste des figures
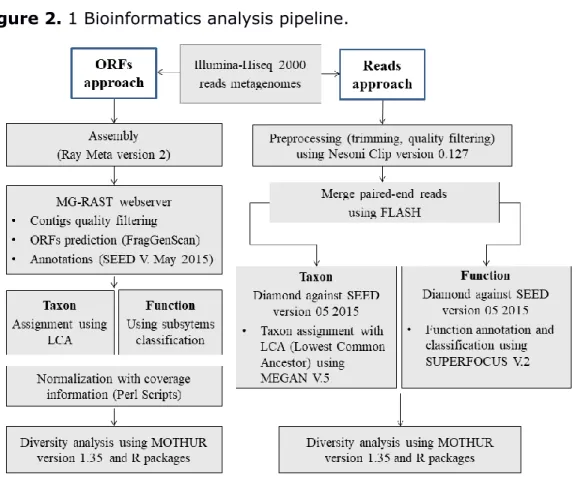
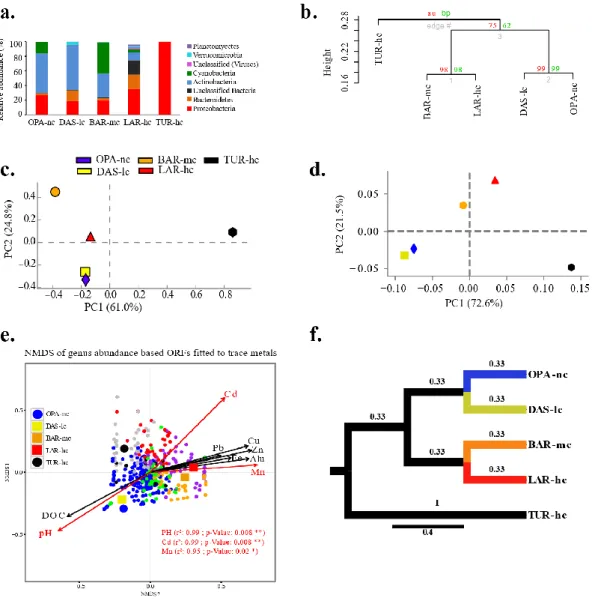
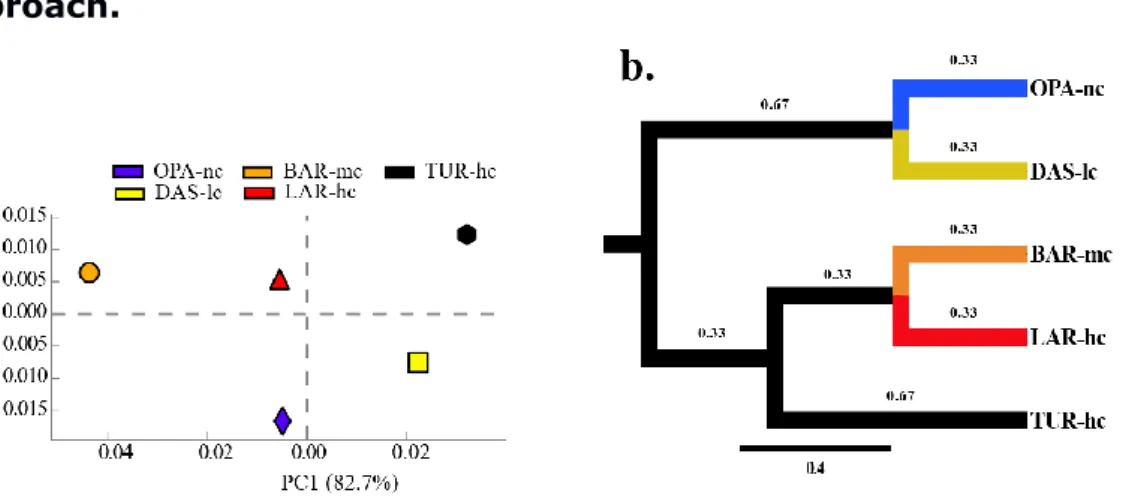
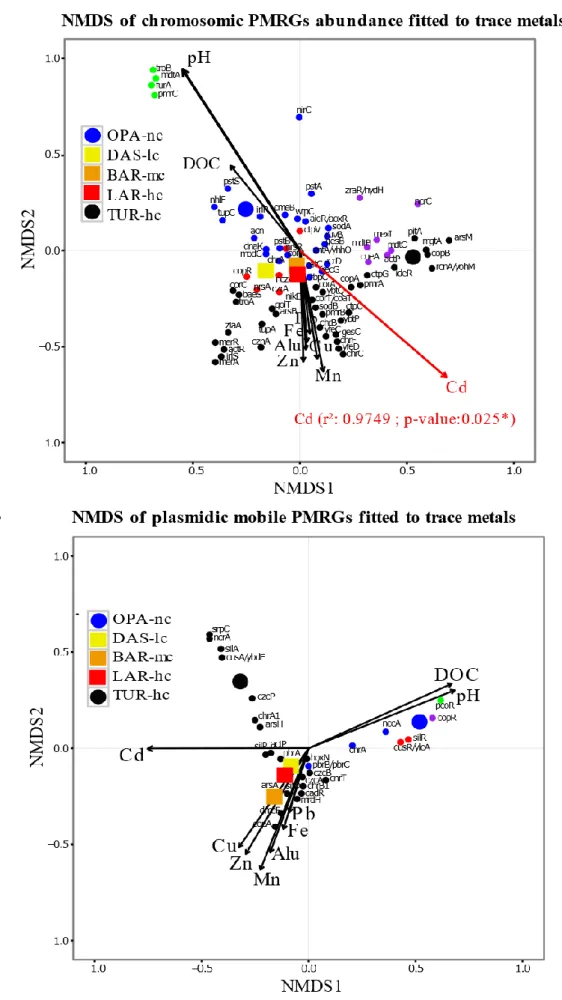
Figure 2. 1 Bioinformatics analysis pipeline. ... 74 Figure 2. 2 Composition of metacommunities based on the ORF approach. 75 Figure 2. 3 Function abundance classification based on ORF approach. ... 76 Figure 2. 4 Polymetallic resistance genes (PMRG) abundance correlation with trace metals. ... 78 Figure 2. 5 Decoupling of taxon and function between metacommunities based on the subsampled reads approach. ... 80 Figure 2. 6 Coupling of taxon and function between metacommunities based on the subsampled reads approach ... 81
Figure 3. 1 Linear variations of alpha-diversity over time and between
treatments explained by the linear mixed model in water and host-microbial communities ... 119 Figure 3. 2 Phylogenetic divergence at the community level ... 120 Figure 3. 3 Correlational co-abundance networks of gut microbial
community. ... 121 Figure 3. 4 Correlational co-abundance networks of skin microbial
community. ... 122 Figure 3. 5 Correlational co-abundance networks of water microbial
community. ... 123 Figure 3. 6 Bar plots of neutral OTUs change at community and
meta-community levels. ... 124
Figure 4. 1 Schematic illustration of the perch microbiome recovery
experiment. ... 158 Figure 4. 2 Alpha-diversity dynamics in the water and perch microbiome. ... 158 Figure 4. 3 Taxonomic composition dynamics of host communities ... 159 Figure 4. 4 Heatmaps of differential abundance among host and water communities ... 159 Figure 4. 5 Function diversity dynamics in host and water microbiome. .. 160 Figure 4. 6 Recovery dynamics of the networks of host communities. .... 161 Figure 4. 7 Recovery dynamics of the network of water communities ... 162 Figure 4. 8 Centrality plots of host microbiome networks ... 163 Figure 4. 9 Percentage of neutral OTUs over time and treatment. ... 164 Figure 4. 10 Demographic variation of metacommunity neutrality across water and host microbiome ... 165
xiii
Liste des abréviations
16S rDNA: 16S Ribosomal DNA;
ACE: Abundance-based coverage estimator; Al: Aluminium;
AMD: Acid Mine Drainage; ANOVA: Analysis of variance;
BAR-mc: Arnoux Bay, medium contaminated; BEB: back extraction buffer;
BH: Benjamini-Hochberg correction test; CC : Cadmium Constant Concentration; Cd : Cadmium;
CdCl2: Cadmium Salts;
CPAUL: Comités de protection des animaux de l’université Laval ; Ctrl: regime of negative Control;
Cu: Cooper;
CV: Cadmium Variable Concentration; DAS-lc: Dasserat Lake, low contaminated; FDR: False Discovery Rate;
Fe: Iron;
HGT: Horizontal Gene Transfer;
LAR-hc: Arnoux Lake, high contaminated; Lead: Pb;
Mn: Manganese;
xiv
NGS: Next generation sequencing; NLS: non-linear least squares model;
NMDS: non-metric Multi-Dimensional Scaling; OPA-nc: Opasatica Lake, not contaminated; ORF: Open Reading Frame;
OTU: Operational taxonomic Unit; PCA: Principal Component Analysis;
PCG: Polymetallic Contamination Gradient; PCR: Polymerase Chain Reaction;
PERMANOVA: Permutational analysis of variance; PMRG: Polymetallic resistance genes;
ppb: parts per billion;
rCCA: Regularized canonical correlation analysis; RDP: Ribosomal Database Project;
TUR-hc: Turcotte Lake, high contaminated; UTO : Unité Taxonomique Opérationnelle; Zinc : Zn;
xv
À Hadi, Amar et Farah À toute ma famille À la révolution de 17 Octobre
xvi
Remerciements
En premier, je présente mes profonds et sincères remerciements à mon directeur Pr. Nicolas Derome de m’avoir donné l’opportunité de continuer dans la recherche scientifique, et la chance de saisir ma première opportunité québécoise. Merci de m’avoir accueilli dans ton laboratoire comme un assistant de recherche en premier temps (Décembre 2013-Mai 2014) et comme un étudiant en doctorat en deuxième temps (Juin 2014-Octobre 2018) ! Merci de m’avoir accordé toute ta confiance pour mener mon projet avec toute liberté ! Merci de m’avoir donné avec toute générosité tous les matériels et les consommables nécessaires pour mener mes travaux dans ton laboratoire ! Merci de m’avoir donné toute cette belle chance.
J’aimerais aussi remercier les membres de mon comité d’encadrement : Pr. Connie Lovejoy, Pr. Louis Bernatchez et Pr. Jacques Corbeil. Merci pour votre confiance aux différentes étapes de ce doctorat et pour vos suggestions. Je voudrais remercier de tout mon cœur Dr. Martin LLewellyn de m’avoir accordé également toute sa confiance pour réaliser des collaborations scientifiques sur ses projets durant son post-doctorat dans notre laboratoire. Merci Martin pour ton soutien tous les niveaux et merci de m’avoir accueilli dans ton laboratoire comme un chercheur postdoctoral à l’université de Glasgow, même avant de soutenir ma thèse.
Merci infiniment à Dr. Mohamed Alburaki de m’avoir soutenu durant cette thèse. Merci Mohamed de m’avoir donné l’opportunité de collaborer sur ton projet et de m’avoir transmis ta passion pour les abeilles, et de me faire connaitre leur diversité génétique en Syrie et au Liban.
Grand merci à tous mes proches collaborateurs, Hamza Seghouani, Sarah El-Khoury, François-Etienne Sylvain, Pierre-Luc Mercier et Dr. Umer Ijaz.
Je tiens à remercier particulièrement Jeff Gauthier d’avoir lu mon texte en Français. Merci pour toutes les discussions infinies et intéressantes sur les aspects différents de la recherche scientifique. Tu es un collègue agréable, altruiste, et toujours prêt pour aider tous les collègues au laboratoire.
xvii
Merci spécial à Amina Abed et Vani Mohit d’avoir corrigé minutieusement plusieurs textes en Français à plusieurs occasions durant mon parcours. Merci à toi Amina pour tes conseils concernant les protocoles de l’extraction d’ADN. Merci à Dr. Anne Dalziel, Dr. Amanda Xuereb, Dr. Ciara Keating, Eleanor Lindsay, et Matt Bywater d’avoir m’aidé dans les corrections de mes manuscrits en anglais.
Merci à tous les anciens et les nouveaux membres du laboratoire Derome, pour leur qualité humaine et leur aide durant des longues journées d’échantillonnage au LARSA. Merci à Émie, Sidki, Laurence, Katherine, Camille et Sara-Jane.
Merci aux professionnels de recherche du Laboratoire Bernatchez, Alysse et Cecilia pour leur dépannage de matériels durant les longues semaines de grève.
Je souhaiterais remercier spécialement Dr. Michel Lavoie pour ses conseils d’or concernant la manipulation de Cadmium. Merci Michel, tu m’avais ouvert les yeux sur des aspects que j’ignorais en Chimie environnementale.
Je tiens à remercier les personnels du LARSA particulièrement Jean-Christophe Therrien, et je n’oublie pas la plateforme de séquençage de l’IBIS particulièrement Dr. Brian Boyle.
À mes collègues co-fondateurs du Club Bioinformatique, Jeff Gauthier, Dr. Anthony Vincent et Éric Normandeau, je vous exprime tous mes sincères remerciements.
Je souhaiterais remercier Mario Boutin une personne qui a laissé une empreinte humaine et un vide éternel à l’IBIS, et aux services de la Laverie. Mario était toujours plein de joie et d’amour pour ses collègues, que ton âme repose en paix.
À tous(tes) mes ami(e)s Nabil, Carlos, Aref, Yazan, Hayan, Mohamed, Fawzi, Aoun, Rabih, Imad, Roba, Sarah, Émilie, Hector sans oublier personne, je souhaiterais vous remercier pour votre encouragement et pour tous les bons moments. Merci de m’avoir soutenu sur place et à distance durant toutes les étapes de ma vie canadienne.
xviii
À ma mère, mon père, mes frères et sœurs, aucun mot ne suffit de vous exprimer ma gratitude, sans vos encouragements, je ne serais jamais arrivé à finir ma thèse.
xix
Avant-propos
Cette thèse de doctorat en biologie et bioinformatique comporte cinq chapitres incluant une introduction et une conclusion générale. Mes travaux sont constitués de trois articles de recherche, identifiés chapitres 2, 3 et 4 dans la table des matières. Les trois chapitres sont rédigés en anglais car le premier fut publié et les deux derniers demeurent en révision dans des revues scientifiques internationales utilisant la langue anglaise. Le thème global de cette thèse s’articule sur l’axe de l’évolution contemporaine de deux modèles systèmes microbiens : environnemental et hôte-associé dans un contexte d’écotoxicologie.
Le 1er chapitre est une revue bibliographique des principes de bases en écologie microbienne. Le deuxième chapitre fait l’objet d’une publication scientifique dans le Journal « Frontiers in Microbiology » et présente les signatures adaptatives détectées au sein d’un système microbien environnemental sous un gradient de pression sélective, induite par une exposition chronique aux métaux traces au cours de plus de 60 ans d’exploitation minière.
Le 3ème et le 4ème chapitres présentent les empreintes de l’évolution expérimentale d’un système microbien hôte-associé dans un contexte d’intoxication métallique artificielle selon deux régimes de sélection, constant et graduel. Le chapitre 3 se focalise sur les signatures de la résistance de microbiote de l’hôte en fonction de l’intensité du polluant, tandis que chapitre 4 décrit la résilience de la structure de microbiote en fonction du gradient de stress métallique. Les deux chapitres sont sous révisions depuis quelques mois, dans les deux revues « Nature ISME » et « Microbiome » respectivement.
Pour chacun des chapitres de ma thèse, j’ai formulé les hypothèses scientifiques et la théorie des questions adressées avec l’appui de mon directeur Nicolas Derome. Pour le deuxième chapitre la prise d’échantillons sur le terrain a été assurée par Pierre-Luc Mercier le troisième co-auteur de la publication. Le deuxième cosignataire, Malo Le Blouch a contribué également dans l’analyse descriptive des données de séquençage. Pour les chapitres 3 et 4, j’ai principalement contribué à la conception, la planification
xx
et à la conduite des expériences avec l’aide de mon directeur Nicolas Derome et mon collaborateur Hamza Seghouani. La prise des échantillons et la dissection des poissons ont été réalisée grâce à l’aide de tous les membres du laboratoire. Après l’échantillonnage, j’ai effectué la filtration de l’eau, l’extraction de l’ADN, les préparations des librairies, les analyses bio-informatiques et biostatistiques des résultats et la rédaction des articles. Dans le chapitre 3, les deux co-auteurs Katherine Vandal-Lenghan et Pierre-Luc Mercier ont contribué à l’extraction de l’ADN de certains échantillons et la quantification des librairies de séquençage.
Les coauteurs internationaux de l’Université de Glasgow, Martin LLewellyn et Umer Ijaz ont contribué dans l’amélioration de l’analyse des résultats et dans la qualité de la rédaction des manuscrits de chapitre 3 et 4.
Les détails des articles publiés ou soumis se trouvent ci-dessous : Article I. Taxon-function decoupling as an adaptive signature of lake
microbial metacommunities under a chronic polymetallic pollution gradient Auteurs : Bachar Cheaib, Malo Le Boulch, Pierre-Luc Mercier, et Nicolas Derome
Publié le 3 Mai 2018 dans le journal Frontiers in Microbiology (Front Microbiol. 2018 ; 9 : 869.).
Article II. From networks to models: the Yellow Perch (Perca flavescens)
microbiome assembly under metal toxicity
Auteurs : Bachar Cheaib, Hamza Seghouani, Martin Llewellyn, Katherine Vandal-Lenghan, Pierre-Luc Mercier, and Nicolas Derome.
Soumis le 05 Mars 2019 dans journal Nature ISME, rejeté uniquement par l’éditeur (accepté par l’arbitre) le 30 septembre 2019 avec l’option de resoumettre une version courte (Reference ISMEJ-19-00341A)
Article III. Community recovery dynamics in yellow perch microbiome after
gradual and constant metallic perturbations
Auteurs : Bachar Cheaib, Hamza Seghouani, Umer Zeeshan Ijaz, and Nicolas Derome.
xxi
Cet article a été publié dans le journal Microbiome. Microbiome 8, 14 (2020) https://doi.org/10.1186/s40168-020-0789-0
En parallèle, je me suis intéressé à l’évolution d’une métalloenzyme le carbonique anhydrase. C’est une famille protéique ubiquitaire capable de substituer les ions métalliques pour assurer la photosynthèse chez les phytoplanctons. J’ai rédigé un manuscrit (Juin 2017) sur ce sujet sous forme d’un chapitre supplémentaire sous la direction de Connie Lovejoy. Ce travail serait bientôt envoyé à une revue scientifique afin de le publier.
D’un autre côté, les travaux de recherche de la thèse ont été communiqué sous forme de présentations et affiches à des journées scientifiques départementales et des organisations scientifiques québécoises et canadiennes, et à des conférences internationales. J’ai personnellement conçu et communiqué chacune de ces présentations et des affiches. Mes participations en personne sont citées ci-dessous :
Conférences locales à l’Université Laval, Québec, Canada
Plusieurs éditions de la journée d’étudiante (2014, 2015, 2016 et 2018) de l’Institut de Biologie Intégrative et des Systèmes (IBIS)
Deux éditions de colloque annuel de département de Biologie (2015,2018) avec un prix de distinction (Edition 2015) de la fondation Richard Bernard.
Conférences et réunions annuelles canadiennes
Plusieurs éditions (2014, 2015, 2017) de la Réunion annuelle des Ressources Aquatiques Québec (RAQ), Québec.
Colloque annuel conjoint RÉAQ-EcoBIM (ÉcoBIM 2015), INRS, Québec. Conférence Genomes to / aux Biomes (2014), 1ère réunion conjointe de la
Société canadienne d'écologie et d'évolution (SCEE), de la Société canadienne de zoologie (CSZ) et de la Société canadienne des limnologie (SCL), 25-29 mai, Montréal.
BISP 2016. Congrès BiSP (Bactériologie intégrative : Symbiose & Pathogenèse), third edition, Université Laval Québec, Canada.
xxii
Le 17ème Symposium de la Société Internationale de l'Écologie Microbienne (ISME17), du 12 - 17 Août 2018, Leipzig, Allemagne. Ma participation été financé en partie par les Fonds général pour les études supérieures (FGES) du Conseil de recherches en sciences naturelles et en génie (CRSNG).
Le 16ème Symposium de la Société Internationale de l'Écologie Microbienne (ISME16), du 21 au 26 août 2016, Montréal, Canada.
GLBIO-CCBC 2016, Great Lakes Bioinformatics et Conférence Canadienne sur la Biologie Computationnelle, du 16 au 19 mai 2016, Toronto, Canada. Mes recherches dans le laboratoire de Nicolas Derome, ne se restreignent pas aux chapitres présentés dans ce document. J’ai contribué en tant que co-auteur aux analyses bioinformatiques et biostatistiques de quatre publications sur des problématiques qui rentrent dans l’intérêt général de cette thèse
La première porte sur l’impact de l’acidité de l’eau sur la résilience de microbiote d’un poisson amazonien le Tambaqui publiée dans le journal
Scientifc Reports
Sylvain, F.-É., Cheaib, B., Llewellyn, M., Gabriel Correia, T., Barros Fagundes, D., Luis Val, A., Derome, N., 2016. pH drop impacts differentially skin and gut microbiota of the Amazonian fish tambaqui (Colossoma macropomum). Sci. Rep. 6, 32032.
La deuxième porte sur la causalité entre pesticides, expression des gènes et maladies infectieuses des abeilles (varroa) publié dans Journal of economic entomology.
Alburaki, M. Cheaib B, et al. Agricultural Landscape and Pesticide Effects on Honey Bees (Hymenoptera: Apidae) Biological Traits. Journal of economic entomology 110, 835–847;2017.
La troisième propose un candidat probiotique pour réduire l’effet du parasitisme sur la mortalité des abeilles, publié dans Frontiers in Ecology and
Evolution
El Khoury S, Rousseau A, Lecoeur A, Cheaib B, Bouslama S, Mercier PL, Demey V, Castex M, Giovenazzo P, Derome N. Deleterious Interaction
xxiii
Between Honeybees (Apis mellifera) and its Microsporidian Intracellular Parasite Nosema ceranae Was Mitigated by Administrating Either Endogenous or Allochthonous Gut Microbiota Strains. Frontiers in Ecology and Evolution. 2018 May 23; 6:58. Doi: 10.3389/fevo.2018.00058.
La quatrième révèle une sélection diversifiante des protéases de surface de
Trypanosoma cruzi GP63 parmi les patients atteints de la maladie de Chagas
chronique et congénitale. Elle est publiée dans le journal PLOS Neglected Tropical Diseases
Llewellyn MS, Messenger LA, Luquetti AO, Garcia AL, Torrico F, Tavares SBN,
Cheaib B, Derome N et al. 2015. Deep sequencing of the Trypanosoma cruzi
GP63 surface proteases reveals diversity and diversifying selection among chronic and congenital Chagas disease patients. PLoS NTD.
Depuis le début de la dernière année de cette thèse, j’entame un stage post-doctoral avec mes collaborateurs, William Sloan, Martin LLewellyn et Umer Ijaz à l’école ingénieure de l’Université de Glasgow. Mes recherches portent sur la modélisation écologique de la dynamique de microbiote des salmonidés. Mes recherches se concentrent sur la quantification du rôle relatif des processus neutres et non-neutres dans la colonisation du tube digestif des juvéniles des salmonidés dans la nature et en aquaculture. Mes résultats pour l’instant sont publiés dans « le journal of AEM Applied and Environmental
Microbiology »
Durant cette thèse, j’ai transformé mes difficultés financières en opportunités pour développer un gout pour l’enseignement en assurant mes fonctions d’auxiliaire d’enseignement de biostatistique sous forme des travaux dirigés avec Frederic Maps. Durant mon post-doc, j’ai également saisi l’opportunité d’assister des travaux dirigés sur les bases de programmation (Python) avec mon collaborateur Umer Ijaz à l’université de Glasgow.
Sur le plan social et scientifique général, j’ai initié l’idée du Club Bioinformatique de l’IBIS que j’ai co-fondé plus tard avec mes chers collègues Jeff Gauthier, Antony Vincent, et Eric Normandeau de plusieurs laboratoires de l’IBIS. Cette expérience m’a offert la chance de partager et d’améliorer mes connaissances et d’acquérir des nouvelles problématiques récentes en bioinformatique biostatistique et en biologie évolutive
1
Introduction Générale
L’anthropocène, soit l’époque géologique contemporaine, est caractérisée par un impact colossal de l’activité humaine sur la biosphère (Balter 2013; Larson et al. 2014; Waters et al. 2016; Tucker et al. 2018). De l’industrie chimique, plastique et métallurgique, aux pesticides et insecticides, à la pollution des eaux, et la perturbation des cycles biogéochimiques de la terre, la liste est longue et ne cesse de s’allonger avec des centaines d’altérations irréversibles des écosystèmes. Les effets anthropiques sur les écosystèmes terrestres, fluviaux et marins et les systèmes biologiques sont devenus alors tout à la fois néfastes et étendus à l’échelle mondiale. Ainsi, le progrès technologique d’Homo sapiens (industriel, numérique, communication, énergétique, transport, nucléaire, militaire, etc.), sa croissance démographique et la prolongation de son espérance de vie ne pourraient plus continuer au détriment des ressources de la biodiversité et de l’équilibre de nos écosystèmes (Crutzen and Stoermer 2000; Pelletier and Coltman 2018). Au-delà des enjeux économiques, politiques de la réalité et en dépit de l’absence des règlementions éthiques efficaces par les nations unies, les chercheurs ne cessent de saisir l’opportunité pour soulever des questions fondamentales concernant l’accélération artificielle de l’évolution génotypique des systèmes biologiques sous l’effet de la pression sélective induite par les effets anthropiques. À court et long terme, les études de l’évaluation de l’impact des xénobiotiques sur la biodiversité tout au long de la chaine trophique démontrent des perturbations même à l’échelle la plus fine du vivant, les micro-organismes ou les microbes. Les xénobiotiques sont des produits chimiques trouvés mais non produits par les organismes ou l'environnement. Certains produits chimiques naturels (endobiotiques) deviennent des xénobiotiques lorsqu'ils sont présents dans l'environnement à des concentrations excessives. Le terme « xeno » dans « xénobiotiques » vient du mot grec « xenos » qui signifie un invité, ami ou étranger (Soucek 2011). Sans métabolisme, de nombreux xénobiotiques atteindraient des concentrations toxiques (Croom 2012).
2
L’anthropocène: des effets anthropiques sur la biodiversité et l’assemblage des communautés microbiennes
Il est connu que les microbes (Bactéries, Archées, Eucaryotes unicellulaires, Virus, etc.) sont ubiquitaires, vivant en communautés, en biofilms, planctoniques ou associées avec d’autres forme de vie. Les communautés microbiennes planctoniques se trouvent dans l’eau, le sol et l’air, et les symbiotiques se sont associées aux Métazoaires et aux Plantes, et aux autres microbes unicellulaires. Elles sont les productrices primaires des services écosystémiques du sol, et de l’eau, ou de l’hôte et constituent la partie majeure de la biodiversité sur terre.
Les communautés microbiennes impliquées dans ces services s’avèrent très impactés dans leurs répertoires génotypiques et phénotypiques par les xénobiotiques. Citons par exemple les antibiotiques, les métaux toxiques, les plastifiants chimiques, les biocides comme les pesticides, insecticides, et désinfectants. Ces polluants chimiques non dégradables sont souvent des xéno-estrogènes synthétiques qui interfèrent avec les récepteurs des systèmes biologiques comme des perturbateurs endocriniens. Dans ce contexte de perturbation, des centaines d’exemples peuvent être cités, nous citons ici quelques-uns.
L’exemple de traitement par des antibiotiques est un excellent argument qui témoigne de l’adaptation rapide des microbes et l’accélération de la cadence de l’évolution microbienne. Les études montrent que les souches bactériennes traitées par des antibiotiques (Vincent et al. 2019) acquièrent de la résistance par conjugaison, un mécanisme de transfert horizontal parmi d’autres (transformation, transduction) bien connus chez les microbes. Cette résistance prépondérante aux antibiotiques est acquise horizontalement et avant de se transmettre verticalement d’une génération bactérienne à l’autre par division clonale (Holmes et al. 2016; von Wintersdorff et al. 2016; Cesare, Eckert, and Corno 2016). Les biocides (triclosan, toluène, méthyl-mercure, proflavine, etc.) sont des polluants de classes chimiques différentes qui conduisent aussi à l’évolution du résistome bactérien (l’ensemble de gènes de résistance bactériens) par des mutations génomiques (substitutions, délétions, insertions) ou par l’acquisition de nouveaux gènes par transfert
3
horizontal. Peu importe les mécanismes, si les mutations génétiques sont fixées dans les populations à travers les générations, elles peuvent selon la stabilité de l’environnement conférer des avantages phénotypiques qui contribuent à des capacités adaptatives comme la dégradation des xénobiotiques. D’autre part, les néonicotinoïdes (Clothianidine, thiaméthoxame ou imidaclopride) affectent la physiologie, le comportement des pollinisateurs, et par conséquent la santé de l’Homme et de notre environnement (Raine 2018; Crall et al. 2018; Alburaki et al. 2016; Doublet et al. 2015; van der Sluijs et al. 2013; Di Prisco et al. 2013). Par exemple, les pesticides et les insecticides contiennent des molécules chimiques qui perturbent non seulement les capacités cognitives (Zhang and Nieh 2015; Siviter et al. 2018) et hygiéniques des abeilles (Boutin et al. 2015; E. Zhang and Nieh 2015; Siviter et al. 2018), mais aussi leur microbiote (Raymann, Shaffer, and Moran 2017; Motta, Raymann, and Moran 2018), ce qui augmente leur susceptibilité aux pathogènes et de ce fait perturbe l’équilibre immunité-symbionte/microbiote.
Le dernier exemple concerne les métaux lourds, ayant une masse atomique élevée, certains laissent de traces toxiques (Cd, Ni, Cu, Co, Al etc.) qui altèrent la biodiversité microbienne dans le sol, les milieux aquatiques et les systèmes biologiques (Koschorreck 2008; Huang, Kuang, and Shu 2016; Hudson-Edwards and Dold 2015). Les études publiées par notre laboratoire ont montré une corrélation entre les concentrations des métaux avec la variation la composition des communautés microbiennes (Laplante and Derome 2011; Laplante, Boutin, and Derome 2013). Par exemple, les métaux lourds déversés par le drainage minier acide, en particulier le cadmium, augmente l’acidité de l’eau, et induit des changements dans la composition taxonomique des communautés bactériennes favorisant les Alphaprotéobactéries.
Les effets des métaux traces toxiques sur l’environnement (Nordstrom 2011; Bejan and Bunce 2015; Lavoie, Fortin, and Campbell 2012; Wu et al. 2016; X. Zeng, Chen, and Zhuang 2015) et les êtres vivants (Vymazal 1987; Giguère et al. 2004; Lacroix and Hontela 2004) sont connus, mais très peu étudiés dans le cas des symbiontes microbiens associées aux systèmes biologiques aquatiques, par exemple les Poissons (S. Zhang et al. 2015;
4 Bridges et al. 2018)
En général, les xénobiotiques perturbent la diversité, la structure et les fonctions des communautés microbiennes, leur assemblage ainsi que leurs interactions, selon des processus moléculaires évolutifs et adaptatifs méconnus.
Les études de l’assemblage du microbiote de l’Homme sous antibiothérapie (Costello et al. 2012) ou du microbiote de poisson euryhalin lors de l’acclimatation à la salinité (V. T. Schmidt et al. 2015), étaient principalement centrées sur des processus déterministes, avec peu de preuves d’une colonisation stochastique. De plus, le rôle des processus écologiques et évolutifs dans la résilience de la structure des communautés microbiennes après perturbation reste à déchiffrer. Théoriquement, la nature de ces processus peut varier entre neutre (stochastique) (Sloan et al. 2006; Jayathilake et al. 2017) et sélective (déterministe) (Stegen et al. 2012; Q. Zeng et al. 2017). Dans le contexte de la résilience du microbiote de l’hôte ou environnemental, peu d’études ont été dédiée à cette question. Ces derniers ont révélé que ce sont les processus déterministes qui induisaient la dynamique de la succession bactérienne étudiée dans un contexte du sol perturbé soit par un gradient d’épuisement des éléments nutritifs (Song et al. 2015), soit par un choc thermique (Jurburg et al. 2017), ou par une réhydratation pluviale(Placella, Brodie, and Firestone 2012)
Néanmoins, il reste encore beaucoup à faire pour comprendre les mécanismes de l’assemblage des communautés microbiennes résilientes chez diverses espèces d’organismes hôtes sous un contexte de perturbation et de résilience.
Les questions et les chapitres de cette thèse
Dans cette thèse, les impacts des effets anthropiques sur la diversité, la structure, la fonction et la composition des communautés microbiennes dans deux écosystèmes ; environnemental, et en association avec un hôte seront discutés. En admettant que les connaissances actuelles sur l’évolution microbienne à partir des microbes cultivables ne soient pas représentatives de celles de la majorité inconnue, les approches de séquençage de nouvelle génération « Next generation sequencing » (NGS) et le progrès de la
bio-5
informatique (banques des séquences, réseaux, algorithmiques), ainsi les modèles récents en écologie et évolution microbienne nous ouvrent alors des voies interdisciplinaires et intégratives prometteuses pour trouver des réponses à nos questions. Ainsi dans cette étude, nous nous sommes intéressés à trois problématiques :
1) La dynamique de la structure et de la composition des communautés microbiennes, et leurs fonctions écosystémiques 2) L’évolution de leurs interactions et leur assemblage dans un
contexte anthropogénique
3) La résilience de leur structure et assemblage et leurs interactions avec leur écosystème après perturbation (environnement et hôte).
Le premier chapitre est une revue bibliographique qui résume les méthodes
et les concepts de base en écologie microbienne.
Le deuxième chapitre étaye l’hypothèse que les perturbations chroniques
des communautés microbiennes lacustres par des métaux traces entraineraient un découplage entre la diversité taxonomique et fonctionnelle. Pour vérifier cette hypothèse, des communautés microbiennes ont été échantillonnées dans cinq lacs exposés à un gradient de contamination polymétallique (PCG), appartenant à un même bassin versant, situé à proximité d’une mine de cuivre historiquement active pendant plus de soixante ans. Avec une approche métagénomique, bio-informatique, nous avons alors caractérisé le niveau d’intégrité des fonctions représentant les services écosystémiques.
Le troisième chapitre étaye l’hypothèse qu’un processus de gradient
sélectif gouvernerait l'assemblage du microbiote d’un organisme soumis à des perturbations graduelles et constantes.
Pour évaluer cette hypothèse, une approche d’évolution expérimentale à court terme a été utilisée (six mois), laquelle, compte tenu du temps de génération des Bactéries (20 à 30 minutes chez E. coli) équivaut à plus de 8000 générations. Plus précisément, nous avons testé expérimentalement au laboratoire comment l’exposition prolongée constante ou graduelle au
6
cadmium (Cd) de 1200 individus des perchaudes (Perca flavescens) avait affecté l’assemblage de leurs microbiotes par séquençage d’amplicons du gène de l’ARNr 16S. Nous avons constaté qu’un gradient de sélection induit par un gradient de concentration d’un métal toxique a perturbé non seulement la physiologie de l’hôte, mais également le recrutement et l’assemblage de son microbiote.
Le quatrième chapitre traite de la dynamique de la résilience du microbiote
de la perchaude après l’arrêt de l’exposition à des quantités sous-létales de cadmium. Le terme « résilience » est employé pour décrire le changement qui se produit lorsqu'une communauté retourne à un autre état stable après perturbation. Le rétablissement des communautés microbiennes dépend du type, de la durée, de l’intensité et du gradient de perturbation.
Nous focalisons sur les modèles car les rôles des processus écologiques et évolutifs dans la résistance et la résilience de la structure du microbiote des hôtes restent à documenter. Théoriquement, et telle que mentionné ci-dessus, la nature de ces processus varie entre neutralisme (stochastique) (Stephen P. Hubbell 2006; Sloan et al. 2006) et sélection (déterministe) (Webb et al. 2002; Chase 2003). Ces derniers opèrent soit par filtrage environnemental et exclusion compétitive (Cadotte et al. 2010; Stegen et al. 2012). Le peu d’études disponibles a révélé que ce sont des processus déterministes qui régissent la dynamique de la succession bactérienne durant le rétablissement des communautés microbiennes des sols. Dans notre étude expérimentale (chapitre 3 et 4), nous avons évalué la contribution relative des processus neutres et non-neutres à la résistance et à la résilience de l’assemblage du microbiote de la perchaude à la suite d’un gradient expérimental d’exposition métallique. Étant donné que les juvéniles de perchaude peuvent tolérer des doses non létales de cadmium sans subir de dommages physiologiques importants (Giguère et al. 2006; Campbell et al. 2005a), notre modèle hôte-microbiote est optimal pour étudier la capacité de résilience du microbiote après une perturbation liée à l'exposition au cadmium. Premièrement, nous avons quantifié avec des méthodes appropriées les niveaux de métaux traces dans des échantillons de foie et d’eau. Ensuite, nous avons comparé l’état de résilience de la structure et de la fonction de la communauté dans l’eau et du microbiote de l’hôte entre des
7
régimes constants et variables d’exposition au cadmium. Afin de démêler l'effet du xénobiotique du développement de l'hôte (Sylvain and Derome 2017; Burns et al. 2016a) sur l'ontogenèse, l'assemblage du microbiote a également été évalué dans des conditions stables en tant que régime témoin. Deuxièmement, nous discutons des changements globaux de la diversité taxonomique et fonctionnelle ainsi que la prévalence des agents pathogènes. Nous évaluons la contribution relative des processus neutres et non-neutres dans la résilience du microbiote de perchaude de deux régimes d’exposition métallique constant et graduel.
8
Chapitre 1 – Méthodes et concepts de
base en écologie microbienne
Ce chapitre est une bibliographie des méthodes et concepts de base utiles pour assurer la bonne compréhension des trois chapitres de recherche de cette thèse.
9
1.1 Les microbes sont ubiquitaires et l’environnement les filtre
Que leur mode de vie soit libre ou associé à un hôte (microbiote) les micro-organismes constituent le générateur fonctionnel des métabolites primaires et secondaires de tout écosystème. Leur importance est donc centrale et primordiale dans les cycles biogéochimiques de la terre, la qualité des eaux, en agriculture (fixation de l’azote, biostimulation de la croissance par solubilisation d’éléments minéraux, biocontrôle des phytopathogènes), en médecine (la médecine personnalisée pour les diabétiques, la médecine régénératrice après transplantation des cellules souches, le traitement des maladies infectieuses par des probiotiques), en industrie agroalimentaire et en sécurité alimentaire, entre autres. Les nouvelles découvertes en microbiologie, en écologie et en évolution microbienne ont, clairement, mis en évidence l’implication de l’environnement à sélectionner ses communautés microbiennes. Un simple exemple très répandu, les nodules racinaires des plantes recrutent les Bactéries bénéfiques fixatrices d’azote (Brill 1975; Dos Santos et al. 2012; Yang et al. 2017). Le système immunitaire de l’Homme joue le rôle du filtre qui retient les bactéries bénéfiques responsables de la maintenance de son homéostasie (Belkaid and Hand 2014; Belkaid and Harrison 2017; Belkaid and Segre 2014). Ainsi, le microbiote contrôle et régule de nombreuses fonctions vitales de l’hôte telles que l’immunité et l’assimilation des nutriments chez les modèles animaux en génétique et en médecine (Rawls, Samuel, and Gordon 2004; Wong and Rawls 2012; Heys et al. 2018; Gould et al. 2018; Wong et al. 2015).
1.2 Accès à la biodiversité microbienne, de la boite de Pétri au métagénome
Les microbes cultivables au laboratoire ne représentent qu’une infime minorité (< 5%) de la biodiversité microbienne (Donachie and Begg 1970; Konopka 1984; Amann, Ludwig, and Schleifer 1995; Connon and Giovannoni 2002; Nichols et al. 2008; Staley and Konopka 1985). Jusqu’à récemment, ceci représentait une contrainte majeure pour caractériser la composition génétique de l’ensemble des communautés microbiennes dans un échantillon d’un environnement donné. Après deux décennies du séquençage du premier génome bactérien (Haemophilus influenzae par Fleischmann et al (1995)
10
(Fleischmann et al. 1995), l’avènement des technologies de séquençage de nouvelle génération Next generation sequencing » (NGS) a favorisé l’accès aux ressources génétiques microbiennes de différents écosystèmes (sol, eau, hôte-symbionte) d’une manière exhaustive. L’ensemble du répertoire génétique d’une communauté microbienne est appelé métagénome. Il faut toutefois garder à l’esprit que les découvertes fascinantes basées sur la minorité cultivable, sans le recours aux approches métagénomiques, ont permis aux biologistes de construire des banques des données généralistes (GenBank, KEGG. UNIPROT etc.) , spécialistes et expertes (SEED, IMG/M, BRENDA, etc.) à l’échelle des fonctions, des domaines, des protéines, de gènes, de génomes et même des populations (Benson et al. 2013; Kanehisa et al. 2014; “UniProt: The Universal Protein Knowledgebase” 2017; Schomburg et al. 2004; I.-M. A. Chen et al. 2019). Ces connaissances acquises contribuent aujourd’hui à donner des réponses claires aux questions centrales portant sur les bases génétiques de la diversité microbienne, de leurs capacités adaptatives et de leurs potentiels évolutifs dans un contexte anthropique, ou naturel.
1.3 L’avènement du séquençage de nouvelle génération
Le NGS (next-generation sequencing) désigne une nouvelle série de technologies qui a révolutionné le séquençage des acides nucléiques et aminés. Le rendement du séquençage a augmenté de manière exponentielle alors que le prix ne cesse de diminuer rendant ces technologies très rentables. Les technologies NGS ont prouvé leur efficacité, rapidité et surtout leur exactitude (Goodwin, McPherson, and McCombie 2016). Plusieurs méthodologies de NGS ont remarquablement influencé nos connaissances dans presque tous les domaines de la biologie moderne, depuis l’évolution moléculaire jusqu’au diagnostic des maladies contagieuses et infectieuses et en passant par les thérapies des maladies génétiques, le champ d’application est très large. Depuis la découverte de la structure de l’ADN en 1953, à l’élucidation du code génétique, cela nous a pris plus d’un quart de siècle pour développer la méthode Sanger, une première génération de séquençage de l’ADN, basée sur une synthèse chimique connue sous le nom « séquençage par synthèse » ; un seul acide nucléique est déterminé à la fois, en fonction de la longueur des séquences d’ADN d’un gène ou d’un génome. Une décennie
11
plus tard, le séquençage Sanger a été automatisé par « Applied Biosystems » en 1987, traçant depuis, la voie vers le séquençage du premier génome bactérien (Haemophilus influenzae) en 1995 (Fleischmann et al. 1995), puis la levure (premier génome eucaryote) en 1996 (Goffeau et al. 1996), et le génome de l’Homme en 2001 (Lander et al. 2001). Ce dernier a coûté 150 millions de dollars (https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost) au consortium international du projet « Human genome » et une longue décennie de cartographie physique et génétique pour compléter le génome. À la fin du projet en 2001, le séquençage de Sanger avait atteint ses limites théoriques et techniques. Par la suite, afin d’accélérer le séquençage, augmenter le rendement et notamment réduire le coût et le temps, une nouvelle ère des méthodes s’ouvra en 2005 avec la technologie 454.
La démocratisation du NGS avait par conséquent réduit drastiquement le coût du séquençage du génome humain de 2.7 milliards ou millions? aux environs 1 000 dollars.
Les méthodes NGS se divise en deux grandes catégories : séquençage par ligation (« SOLiD », « Complete Genomics ») et séquençage par synthèse (Illumina, 454, Ion Torrent). La plupart des technologies se base donc sur la dernière, et se classifie en deux méthodes, le séquençage par clôture cyclique (Illumina), et le séquençage par addition successive des nucléotides (454,
Ion Torrent). En seconde et en troisième générations le séquençage par
synthèse produit respectivement des courtes et des longues lectures (Goodwin, McPherson, and McCombie 2016). Les technologies de courtes séquences (Illumina, Qiagen, 454, Ion Torrent) se caractérisent par les coûts les plus bas, et produisent des séquences de haute précision, utiles pour la détection des variants génétiques à l’échelle populationnelle. Cependant, les technologies de longues séquences (PacBio et ONT) assurent des longues séquences afin d’assembler des génomes complets ou pour d’autres applications comme le séquençage des isoformes. Ainsi, chaque technologie présente ses avantages et ses limites en termes de précision, de fiabilité, de longueur des séquences et d’erreurs de séquençage (Goodwin, McPherson, and McCombie 2016; Kumar, Cowley, and Davis 2019; Vincent et al. 2017).
12
Par conséquent, les méthodes NGS procurent un accès aux ressources génétiques de la biodiversité microbienne. Depuis plus qu’une décennie, elles ont ouvert le chemin aux approches omiques « Omics », dont la métagénomique, pour identifier les micro-organismes et caractériser leur contenu génétique à l’échelle phénotypique, fonctionnelle et métabolique.
1.4 Les approches de la métagénomique
Tout d’abord un métagénome c’est l’ensemble des matériels génétiques des microorganismes séquencés dans un échantillon d’un environnement donné (sol, air, eau, animal hôte, plante hôte).
Les méthodes métagénomiques sont souvent subdivisées en deux approches. La première consiste en l'amplification ou séquençage d’un seul gène marqueur conservé universellement chez tous les micro-organismes, par exemple le gène de la sous-unité 16S de l’ARN ribosomique chez les Bactéries, de la sous-unité 18S chez les Eucaryotes microbiens (zooplanctons et phytoplanctons). Alors que la deuxième approche consiste en un séquençage de l'ensemble du contenu génétique d’une communauté microbienne « Whole-Genome-Sequencing » (WGS), ce qui procure un aperçu global du répertoire fonctionnel et du contenu génétique du métagénome en question.
1.5 L’approche d’amplicons basée sur un gène marqueur universel
Cette approche consiste à amplifier un gène marqueur à partir d’un échantillon d’ADN. Une amplification « PCR » est réalisée pour quantifier l'abondance relative des variants du marqueur en question au sein de la communauté microbienne présente dans un échantillon donné. Le gène ribosomique 16S chez les Procaryotes et le gène 18S chez les Eucaryotes sont donc des marqueurs universels couramment utilisés en phylogénie moléculaire et plus généralement pour détecter les changements de composition des communautés microbiennes.
Le gène de l’ARN ribosomique 16S a été proposé comme un marqueur de phylogénie pour la première fois par Carl Woese (Woese 1987) . La taille de ce gène fait environ 1500 paires de bases, il possède des propriétés qui répondent aux critères d’un candidat d’un marqueur génétique universel pour différencier les différentes lignées bactériennes. Le gène 16S est ubiquitaire,
13
très conservé entre les différentes lignées bactériennes et évolue lentement (Gillespie et al. 2006; Pei et al. 2010). Il est composé de de neuf domaines qui se subdivisent entre deux types de séquences conservées et variables. Les domaines conservés évoluent très lentement avec peu de mutations fixées. Les régions variables sont ciblées par des amorces universelles car elles permettent généralement distinguer entre les différents genres bactériens (Baker, Smith, and Cowan 2003; Guo et al. 2013; Kembel et al. 2012; Caporaso et al. 2011), même si certaines souches peuvent être autant plus divergentes au sein d’un même genre que d’entre genres différents (Johansen et al. 2017; Acinas et al. 2004; Janda and Abbott 2007).
L’amplification de la séquence entière du marqueur n’est pas obligatoirement nécessaire, cependant une région hypervariable serait ciblée comme un code-barre génétique. Après amplification de ce dernier, le produit de PCR est purifié et enfin séquencé. Ensuite, les séquences lues des amplicons sont analysées par des méthodes bio-informatiques, qui se résument par les étapes suivantes :
Le contrôle de qualité consiste sur l'élimination de courtes séquences, la filtration des homopolymères et des séquences ayant un score de qualité moyen à très bas.
La correction du nombre de copies s’il s’agit du marqueur (16S/18S), et filtration des chimères produites par le biais d'hybridation non spécifique des amorces durant l'amplification PCR.
La classification des séquences filtrées en unités de base de diversité tout en se basant sur un seuil de similarité nucléotidique (97% à 99%) arbitraire communément utilisé en écologie microbienne, et à la fois contesté (Edgar 2018). Au sein d’une même unité de diversité obtenue avec une similarité de 97%, les études documentent une grande diversité des espèces, par exemple au sein du genre Bacillus (Maughan and Van der Auwera 2011; Connor et al. 2010), d’où la contestation de l’approche de seuil de similarité. Les unités taxonomiques sont alors annotées taxonomiquement et comparées phylogénétiquement afin de comprendre la structure et la composition des communautés microbiennes en question (Figure 1).
14
1.6 L’approche métagénomique globale basée sur le séquençage de l’ADN total.
Pour cette approche, l’échantillon d’ADN est fragmenté aléatoirement par fragmentation mécanique. Les fragments obtenus sont de différentes tailles, ils sont clonés aléatoirement dans des vecteurs afin de construire une banque de librairies à petits ou larges inserts. Tout dépend de l’intérêt de l’étude, les librairies peuvent être criblées afin de cibler un gène d'intérêt biomarqueur de l’écosystème en question, ou bien séquencées dans son intégralité afin de permettre la caractérisation du contenu génétique et taxonomique du métagénome (Schéma 1.1). Après le séquençage à haut débit, l’assemblage des séquences en « contigs », suffisamment longs permet de prédire le répertoire des gènes dans l’échantillon. Selon la complexité de la diversité de l’environnement étudié, les assembleurs peuvent permettre la reconstitution de génomes presque complets à partir d’un métagénome (Albertsen et al. 2013; Parks et al. 2015; Iverson et al. 2012; Albertsen et al. 2013; Mehrshad et al. 2016). L’analyse bio-informatique des méta-communautés post-assemblage se résume en trois étapes principales :
Prédire des cadres de lecture ouverts (« Open Reading Frame » ORF) des gènes.
Assigner des affiliations taxonomiques de gènes prédits.
Annoter les fonctions de gènes en spécifiant les ontologies de leurs fonctions. L’abondance de chaque gène est donc déterminée par la fréquence de son occurrence et la profondeur des séquences lues qui ont servi à l’assemblage de son contig prédit.
15
Schéma 1. 1 Approches métagenomiques de l’analyse de microbiote.
Édité de Morgan XC, Huttenhower C (2012) PLoS Comput Biol 8(12): e1002808.doi:10.1371/journal.pcbi.1002808
Une étape cruciale de toutes les études en écologie microbienne est le regroupement des séquences des communautés de micro-organismes en groupes (clusters en anglais) phylogénétiquement proches (97% de similarité) appelés OTU (« operational taxonomic unit ») (Schéma 1.1). Cependant, cette approche a été remise en question à plusieurs reprises, car le concept OTU ne prend pas en compte la théorie de spéciation chez les Bactéries (Preheim et al. 2013; T. S. B. Schmidt, Rodrigues, and Mering 2014). Des approches alternatives ont été proposé récemment pour minimiser le biais introduit par le seuil d’identité.
16
La notion de l’OTU représente donc ici l’unité de base de diversité même si le concept derrière la définition de l’espèce demeure problématique et un sujet de débat actif (Doolittle and Zhaxybayeva 2009).
1.8 L’approche des variants des séquences d’amplicons (VSA)
Pour pallier le problème du seuil d’identité (97%) arbitraire de l’approche UTO, il a été proposé récemment que les taxons devraient être définis avec une approche d’identité exacte des séquences des gènes marqueurs (Callahan et al. 2016). Cette approche alternative à l’UTO est basée sur la détermination des taxons par des variants des séquences exacts (VSEs), et connue sous le terme de variants des séquences d’amplicons (VSA)s « ASVs :
amplicons sequence variants » (Callahan, McMurdie, and Holmes 2017) ou
UTO-à-rayon zéro (zUTOs), tel que proposé par Edgar (2016).
Les VSAs de novo sont déduits d'un processus d’apprentissage automatique dans lequel les séquences biologiques sont distinguées des erreurs sur la base, d’un modèle d’erreur via un algorithme de débruitage « Denoising » DADA2 (Callahan et al. 2016) et d’une fonction (f) de transition de probabilité des erreurs. Le modèle d’erreur assume que les séquences répétées ont plus de chances d'être observées que celles contenant des erreurs.
Par conséquent, l'inférence des VSAs de chaque échantillon ne peut pas être effectuée indépendamment de chaque lecture considérée comme la plus petite sous unité de données. Selon Callahan et al. (2017), contrairement aux unités UTOs de novo, les VSAs sont des étiquettes cohérentes car ils représentent une réalité biologique. Ces unités peuvent être déduites et comparés avec différentes études ou différents échantillons. Cette approche n’augmente pas seulement la résolution taxonomique, mais elle simplifie également les comparaisons entre les études en éliminant la nécessité de réannoter les taxons lorsque les ensembles de données sont fusionnés (Glassman and Martiny 2018). Grâce à ces avantages, il y a eu une augmentation du nombre de pipelines bioinformatiques cherchant à utiliser les VSEs et à minimiser le biais d’inférence de la diversité (Callahan, McMurdie, and Holmes 2017; Edgar 2016; Amir et al. 2017). En outre, les auteurs de cette approche ont déclaré que les VSEs devraient remplacer les