T
T
H
H
È
È
S
S
E
E
En vue de l'obtention du
D
D
O
O
C
C
T
T
O
O
R
R
A
A
T
T
D
D
E
E
L
L
’
’
U
U
N
N
I
I
V
V
E
E
R
R
S
S
I
I
T
T
É
É
D
D
E
E
T
T
O
O
U
U
L
L
O
O
U
U
S
S
E
E
Délivré par l'Université Toulouse III - Paul SabatierDiscipline ou spécialité : Physiopathologie
JURY
Dr Jacques BERTOGLIO, rapporteur Pr Alain VERLOES, rapporteur Dr Béatrice PARFAIT, examinatrice
Dr Armelle YART, invitée Pr Maithé TAUBER, invitée Dr Patrick RAYNAL, directeur de thèse Pr Jean-Pierre SALLES, directeur de thèse
Ecole doctorale : Biologie-Santé-Biotechnologies de Toulouse Unité de recherche : unité INSERM U563
Directeur(s) de Thèse : Patrick RAYNAL et Jean-Pierre SALLES Rapporteurs : Jacques BERTOGLIO et Alain VERLOES Présentée et soutenue par EDOUARD Thomas
Le 11 décembre 2009
Titre : Impact sur la signalisation cellulaire des mutations de la tyrosine phosphatase
Remerciements
Les travaux présentés dans ce mémoire de doctorat ont été menés dans l’unité INSERM U563 du centre de Physiopathologie Toulouse-Purpan, dans l’équipe médiateurs et signaux lipidiques, prolifération et différenciation cellulaire dirigée par Jean-Pierre Salles et Patrick Raynal.
Tout d’abord, je tiens à remercier mes deux directeurs de Thèse, Patrick Raynal et Jean-Pierre Salles, pour m’avoir accueilli au sein de leur équipe et avoir dirigé ce travail.
Mes remerciements vont également à Monsieur Alain Verloes, professeur de Génétique et directeur de recherche (unité INSERM U676) à l’hôpital Robert Debré, et Monsieur Jacques Bertoglio, directeur de recherche (unité INSERM U461) à la faculté Paris XI, pour m'avoir fait l'honneur d'être rapporteur de ce travail.
Je remercie aussi Madame Béatrice Parfait, généticienne à l’hôpital Beaujon, Madame Maithé Tauber, professeur de Pédiatrie à Toulouse, et Armelle Yart, chargée de recherche à l’unité INSERM U563, qui ont bien voulu participer au jury.
J’exprime toute ma reconnaissance à Madame Maithé Tauber qui m’a soutenu tout au long de ma formation médicale et scientifique sur Toulouse et m’a encouragé à réaliser une thèse de Sciences.
Je remercie chaleureusement toute l’équipe du Centre de Recherche sur la Croissance (CRC), Françoise Conte-Auriol, Marianne Mus, Arnaud Bros et Sarah Laurencin, pour m’avoir initié à la recherche. Ils auront grandement participé à l’augmentation de ma culture scientifique et générale.
Je tiens également à remercier Armelle Yart pour sa grande disponibilité, ses conseils et son aide précieuse pour la rédaction de l’article et la correction du manuscrit de thèse. J’ai beaucoup apprécié ses qualités pédagogiques et ses connaissances musicales.
Je remercie Jean-Philippe Combier pour son aide majeure dans la réalisation de ce projet. Il m’aura ouvert les yeux aux beautés de la biologie moléculaire.
Un grand merci à Alexandra Montagner, Marie Dance et Carla Sampaio qui ont initié ce travail et Audrey Nédélec qui va le continuer.
Enfin, que l'ensemble des techniciens, ingénieurs, étudiants ainsi que le personnel du secrétariat, soient aussi remerciés pour l'atmosphère agréable qu'ils contribuent à maintenir au sein du département.
Impact sur la signalisation cellulaire des mutations de la tyrosine
phosphatase Shp2 associées aux syndromes de Noonan et LEOPARD
Résumé
Le syndrome de Noonan (SN) est une maladie génétique autosomique dominante relativement fréquente (≈1/2000), caractérisée par l’association d’une dysmorphie faciale, d’un retard statural et d’une cardiopathie. Le syndrome LEOPARD (SL) est une maladie génétique plus rare, phénotypiquement très proche du SN, s’en distinguant essentiellement par l’existence d’une surdité et d’anomalies cutanées. Ces deux syndromes appartiennent à la famille des syndromes « Neuro-Cardio-Facio-Cutanés », un groupe de maladies du développement en rapport avec des mutations germinales de gènes codant pour des molécules impliquées dans la voie Ras/Mitogen Activated Protein Kinase (MAPK).
Au moins 50% des patients SN et plus de 80% des patients SL ont des mutations germinales du gène PTPN11, codant pour la tyrosine phosphatase Shp2. De manière intéressante, les études biochimiques ont montré que les mutations de PTPN11 ont des effets opposés sur l’activité de la phosphatase, gain de fonction dans le SN et perte de fonction dans le SL. Comment des mutations aux effets opposés peuvent être à l’origine de phénotypes similaires est à ce jour incompris.
Un point important dans la compréhension de la physiopathologie des SN et SL est l’identification des voies de signalisation altérées par les mutants de Shp2. Jusqu’ici, la plupart des études se sont focalisées sur la voie Ras/MAPK. Ainsi, les mutants SN de Shp2 seraient responsables d’une activation de cette voie suivant des mécanismes moléculaires peu connus. En ce qui concerne le SL, nos connaissances sont plus limitées car même l’effet des mutants SL de Shp2 sur la voie Ras/MAPK est toujours débattu. Pour aller plus loin dans la physiopathologie de ces syndromes, il nous a semblé important d’étudier l’impact des mutants de Shp2 sur d’autres voies de signalisation, notamment la voie de signalisation PI3K/Akt. Pour cette étude, nous avons travailler à partir de cultures de fibroblastes cutanés immortalisés de patient NS et SL ce qui constituait un outil original et unique à ce jour.
Ce travail nous a permis d’observer, qu’en réponse à l’EGF, la voie PI3K/Akt est hyperactivée dans les cellules de patients SL comparées à celles des patients SN ou des contrôles. Cet effet dominant positif des mutants SL de Shp2 est du à une diminution de la déphosphorylation des sites de liaison de PI3K sur Gab1, augmentant ainsi l’association PI3K/Gab1 et l’activation de la voie PI3K/Akt. Nous avons également observés dans les cellules de patients SL que cette hyperactivation de PI3K/Akt entraîne une augmentation de l’inactivation de GSK3-β et par conséquent une régulation positive des marqueurs d’hypertrophie cardiaque. Enfin, les inhibiteurs de PI3K annulent la croissance hypertrophique induite par les mutants SL dans des cardiomyocytes en culture et des explants cardiaques d’embryons de poulet, suggérant que l’hyperactivation de PI3K/Akt pourrait participer à l’hypertrophie cardiaque souvent observée dans le SL. Ces données expérimentales sont concordantes avec les données cliniques qui ont montré que, dans le cadre des mutations de PTPN11, la fréquence des HCM est élevée (≈50%) chez les patients SL, et basse (<10%) dans le SN.
Il s’agit donc de la première étude suggérant l’implication de la voie PI3K/Akt dans la physiopathologie des SN et SL. Ces données obtenues in vitro restent bien sur à valider, notamment dans des modèles animaux, mais ouvrent de nouvelles perspectives de recherche.
Abréviations
CFC Cardio-Facio-Cutané
CIA Communication inter-auriculaire
CIV Communication inter-ventriculaire
EGF Epidermal Growth Factor
Erk Extracellular-regulated kinase
FAK Focal Adhesion Kinase
FGF Fibroblast Growth Factor
Gab1 Grb2-associated Binder 1
GAP GTPase Activating Protein
GEF Guanosine Exchange Factor
GDP Guanosine Diphosphate
GH Growth hormone
GTP Guanosine Triphosphate
HCM Hypertrophic cardiomyopathy
IGF-I Insulin-like Growth Factor-I
JAK Janus Kinase
JNK c-Jun Terminal Kinase
LMMJ Leucémie myélomonocytaire juvénile
MAPK Mitogen-Activated Protein Kinase
Mek MAPK Erk Kinase
NCFC Neuro-Cardio-Facio-Cutané
NF1 Neurofibromatose de type 1
PDGF Platelet-Derived Growth Factor
PH Pleckstrin Homology
PI3K Phosphoinositide 3-Kinase
PTEN Phosphatase and Tensin Homolog Deleted on chromosome 10
PTK Protein Tyrosine Kinase
PTP Protein Tyrosine Phosphatase
RTK Récepteur à activité Tyrosine Kinase
SC Syndrome de Costello
SH2 Src Homology 2
Shp2 SH2 domain -containing tyrosine phosphatase
SFK Src Family Kinase
SL Syndrome de Noonan
SN Syndrome de LEOPARD
Sos Son of Sevenless
Sommaire
Introduction bibliographique
……….... 1Chapitre I: Description clinique du syndrome de Noonan et des syndromes apparentés 1. Le syndrome de Noonan... 2
1.1. Généralités………..………... 2
1.2. Anomalies de la croissance et de la puberté……… 4
1.2.1. Données cliniques………... 4
1.2.2. Données hormonales……….. 5
1.2.3. Réponse au traitement par GH recombinante……….. 6
1.2.4. Hypothèses physiopathologiques du retard statural……… 8
1.3. Cardiopathie……….. 8
1.4. Anomalies hématologiques et de l’hémostase………... 9
1.5. Anomalies du développement psychomoteur... 10
1.6. Autres signes... 10
2. Le syndrome LEOPARD……… ………… 12
2.1. Anomalies de la croissance et de la puberté……….. 12
2.2. Cardiopathies……….. 13
2.3. Autres anomalies……… 13
3. Les autres syndromes apparentés……… 14
3.3. Le syndrome Cardio-Facio-Cutané……… 14
3.4. Le syndrome de Costello……… 15
3.5. La neurofibromatose de type 1 et le syndrome de Legius………. 16
Chapitre II : Génétique du syndrome de Noonan et des syndromes apparentés : les anomalies de la voie de signalisation Ras/MAPK 1. La voie Ras/MAPK………...……….... 18
1.1. Généralités……….. 18
1.2. Description des molécules impliquées dans la voie Ras/MAPK……… 20
1.2.1. Ras et ses régulateurs……… 20
1.2.1.1. Structure et fonction de Ras………. 20
1.2.1.2. Activation de Ras par la GEF Sos1……… 22
1.2.1.3. Régulation négative de Ras par les GAPs RasGAP et Neurofibromine.. 23
1.2.2. La cascade de signalisation Raf/Mek/Erk……… 23
1.2.2.1. Raf... 23
1.2.2.2. Mek et Erk 1 et 2……… 25
1.3. Régulation de l’activation de la voie Ras/MAPK……….. 26
1.3.1. La tyrosine phosphatase Shp2……….. 26
1.3.2. Les protéines Sprouty et Spred……….. 26
2. Les anomalies génétiques de la voie Ras/MAPK……… 28
2.1. Gènes impliqués dans les syndromes Neuro-Cardio-Facio-Cutanés………. 29
2.1.1. Le gène PTPN11……… 29
2.1.2. Le gène SOS1……… 31
2.1.3. Le gène RAF………... 32
2.1.4. Les gènes MEK 1 et 2... 32
2.1.5. Le gène RAS………. 33
2.1.6. Le gène NF1……….. 35
2.1.7. Le gène SPRED1……….. 36
2.2. Les autres anomalies constitutives de la voie Ras/MAPK……… 36
2.2.1. RASA1 et le syndrome malformation artérioveineuse/malformation capillaire…… 36
2.2.2. NRAS et le syndrome lymphoprolifératif autoimmun……….. 37
Chapitre III :
Corrélations phénotype/génotype dans les syndromes Neuro-cardio-facio-cutanés
1. Syndrome de Noonan………...……… 38 1.1. Retard statural……….……….………. 39 1.2. Cardiopathie……….………. 41 1.3. Anomalies hématologiques……….……….. 42 1.4. Autres atteintes……….……….. 42 2. Syndrome LEOPARD…….……….. 43 2.1. Retard statural………. 43 2.2. Cardiopathie……….. 43 2.3. Autres atteintes………. 43 3. Syndrome Cardio-facio-cutané……… 44 4. Syndrome de Costello……… 46
Chapitre IV : Conséquences fonctionnelles des mutants de Shp2 1. Structure et fonctions biologiques de Shp2……… 48
1.1. Tyrosines kinases et tyrosines phosphatases………. 48
1.2. Structure et régulation de l’activité de Shp2……….. 49
1.3. Gab1, une protéine adaptatrice essentielle à la fonction de Shp2………... 50
1.4. Rôle dans les différentes voies de signalisation……….. 53
1.4.1. Régulation de Ras et PI3K en aval des RTKs……….. 53
1.4.1.1. Régulation de Ras………. 53
1.4.1.2. Régulation de PI3K……….. 55
1.4.2. Régulation de Rho en aval des intégrines……….. 55
1.4.3. Régulation de JAK/STAT en aval des récepteurs aux cytokines……….. 56
1.5. Rôle de Shp2 dans le développement………. 57
1.5.1. Chez les invertébrés……….. 57
1.5.2. Chez les vertébrés………. 58
2. Conséquences fonctionnelles des mutants de Shp2………. 60
2.1. Mutants SN de Shp2………. 60
2.1.1. Conséquences sur l’activité de la phosphatase………. 60
2.1.2. Conséquences sur les voies de signalisation………. 62
2.1.3. Conséquences sur le développement : les modèles animaux……… 62
2.1.3.1. La Drosophile……….. 63
2.1.3.1. Les modèles murins………. 64
Résultats expérimentaux et perspectives
……… 67 Résultats expérimentaux……… 68 1. Introduction……… 68 2. Résultats et discussion……….. 69 3. Conclusion……….. 109 Perspectives……… 1101. Validation du modèle de développement de l’HCM chez les patients SL dans des tissus cardiaques humains et/ou des modèles animaux……….. 110
2. Implication physiopathologique des mutants de Shp2 dans le retard statural... 112
2.1. Hyperactivation de la voie Ras/MAPK sous GH et déficit en IGF-I……….... 112
2.2. Atténuation du retard statural chez les SL par hyperactivation de la voie PI3K/Akt……….... 114
3. Effet des mutants de Shp2 sur différentes voies de signalisation………... 115
4. Conclusion………... 117
Références bibliographiques
………...…. 118Annexes
………... 134Annexe 1 : The Shp2 paradox in Noonan and LEOPARD syndromes: How mutations oppositely influencing its biochemical activity result in similar phenotypes?.. 135
Chapitre I
Description clinique du syndrome de Noonan et des syndromes apparentés
Le syndrome de Noonan (SN, OMIM 163950), décrit par Jacqueline Noonan en 1968 (Noonan 1968), est caractérisé par l’association d’une dysmorphie faciale caractéristique, d’un retard statural et d’une cardiopathie (essentiellement une sténose des valves pulmonaires) (Allanson 1987). Ces caractéristiques cliniques sont partagées à des degrés variables par d’autres syndromes phénotypiquement très proches que sont le syndrome LEOPARD (SL, OMIM 151100), le syndrome Cardio-Facio-Cutané (CFC, OMIM 115150), le syndrome de Costello (SC, OMIM 218040), et la neurofibromatose de type 1 (NF1, OMIM 162200). Nous décrirons dans ce premier chapitre la présentation clinique de ces différents syndromes.
1. Le syndrome de Noonan
1.1. Généralités
Le SN est une maladie génétique relativement fréquente dont la prévalence est estimée entre 1/1000 à 1/2500 naissances vivantes (Nora, Nora et al. 1974; Mendez and Opitz 1985; Opitz 1985; Allanson 1987). Cependant, ces données ne reposent pas sur des études de population précises. De plus, l’incidence est sans doute plus importante du fait d’interruptions spontanées de grossesse.
Le SN peut-être familial avec une transmission le plus souvent autosomique dominante (Jongmans, Otten et al. 2004); cependant, une possible transmission récessive a été suggérée dans certains cas (van Der Burgt and Brunner 2000). Dans ces formes transmises, il existe une prédominance de transmission maternelle (70%) possiblement expliquée par une diminution de la fertilité masculine (Collins and Turner 1973; Nora, Nora et al. 1974; Sharland, Burch et al. 1992). Au moins la moitié des cas sont sporadiques (Nora, Nora et al. 1974; Mendez and Opitz 1985; Allanson 1987; Ranke, Heidemann et al. 1988; Sharland, Burch et al. 1992).
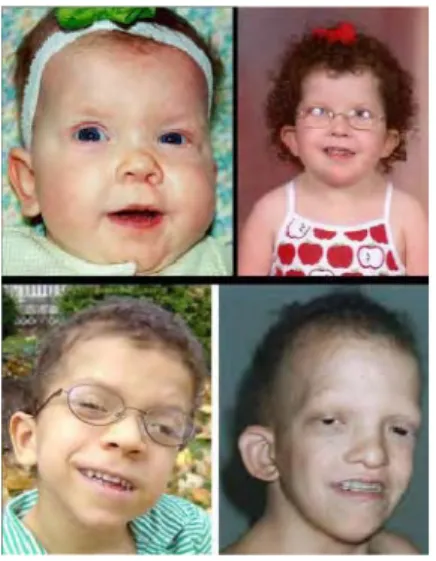
Le phénotype du SN est très hétérogène entre les personnes atteintes, même à l’intérieur d’une même famille. De plus, le phénotype varie au cours des différents âges et peut être discret à l’âge adulte notamment au niveau des signes faciaux (figure 1) (Allanson, Hall et al. 1985; Allanson 1987).
Figure 1. Evolution du phénotype du syndrome de Noonan avec l’âge : « The changing phenotype »
Chez le nouveau-né, les principaux signes sont un hypertélorisme avec des fentes palpébrales tombantes (95%), des oreilles basses en rotation postérieure avec un hélix épais (90%), un philtrum marqué (95%), un palais haut et arché (45%), une micrognathie (25%) et un excès de peau nuccal avec une implantation basse des cheveux (55%). Chez le nourrisson, la tête apparaît relativement large avec turicéphalie, les yeux sont proéminents avec un hypertélorisme et des paupières épaisses « en capuchon ». Le nez a une racine déprimée, une base large et une extrémité bulbeuse. Chez l’enfant plus âgé, le visage apparaît grossier et myopathique, le contour du visage devient plus triangulaire avec l’âge avec la croissance du menton. Il peut exister un ptosis (uni ou bilatéral) et un comblement supraorbitaire latéral. Chez l’adolescent, et l’adulte jeune, les yeux sont moins proéminents et le nez a une racine pincée, un pont haut et fin et une base large. Le cou s’allonge accentuant la palmure et la proéminence du trapèze (90%). Chez l’adulte plus âgé, les plus nasolabiaux sont marqués, l’implantation antérieure des cheveux est haute, la peau transparent et ridée. Les cheveux peuvent être minces chez le nourrisson alors qu’ils sont souvent laineux et bouclés chez l’enfant plus âgé et l’adolescent. Les oreilles basses en rotation postérieure avec un hélix épais sont présentes à tous les âges (Allanson, Hall et al. 1985; Allanson 1987).
Ainsi, établir le diagnostic de SN peut être difficile, spécialement dans les phénotypes modérés et chez les patients adultes. Plusieurs systèmes de scores basés sur les caractéristiques phénotypiques ont été développés pour aider le diagnostic. En 1981, Duncan et al. proposèrent un score diagnostique avec 26 items, difficilement utilisable en pratique
clinique (Duncan, Fowler et al. 1981). En 1993, Sharland et al. définirent comme certain le diagnostic de SN si des caractéristiques faciales typiques (définies dans l’article d’Allanson et al.) étaient associées à au moins une anomalie cardiaque, une petite taille (<10ème percentile) ou une ectopie testiculaire chez le garçon (Sharland, Morgan et al. 1993). Cependant ce score ne permettait pas de diagnostiquer les patients avec une dysmorphie modérée. En 1994, Van Der Burgt et al. développèrent un score plus simple prenant en compte l’importance des modifications faciales (typiques ou évocatrices) (tableau 1) (van der Burgt, Berends et al. 1994). Lorsque ce score clinique est utilisé, le pourcentage de mutations positives est plus important (Jongmans, Sistermans et al. 2005).
retard mental ou cryptorchidie ou dysplasie lymphatique retard mental et cryptorchidie et
dysplasie lymphatique 6. Autres 1er degré dc évocateur 1er degré dc certain 5. Histoire familiale thorax large pectus carinatus/excavatus 4. Anomalie thorax <10èmepercentile <3èmepercentile (-2 DS) 3. Retard statural autres sténose pulmonaire, HCM, anlie ECG
2. Cardiopathie évocatrice typique 1. Dysmorphie B. mineurs A. majeurs Signes
retard mental ou cryptorchidie ou dysplasie lymphatique retard mental et cryptorchidie et
dysplasie lymphatique 6. Autres 1er degré dc évocateur 1er degré dc certain 5. Histoire familiale thorax large pectus carinatus/excavatus 4. Anomalie thorax <10èmepercentile <3èmepercentile (-2 DS) 3. Retard statural autres sténose pulmonaire, HCM, anlie ECG
2. Cardiopathie évocatrice typique 1. Dysmorphie B. mineurs A. majeurs Signes
Tableau 1. Critères diagnostiques du syndrome de Noonan (van der Burgt, Berends et al. 1994)
Le diagnostic de syndrome de Noonan est porté si le patient présente :
Soit une dysmorphie typique (1A) + un signe de 2A à 6A ou 2 signes de 2B à 6B Soit une dysmorphie évocatrice (1B) + deux signes de 2A à 6A ou 3 signes de 2B à 6B
1.2. Anomalies de la croissance et de la puberté
1.2.1. Données cliniques
Le retard statural est présent chez environ 80% des enfants SN et plusieurs auteurs
ont pu établir des courbes de croissance spécifiques à partir de larges séries (Witt, Keena et al. 1986; Ranke, Heidemann et al. 1988).
Chez les enfants SN, les mensurations à la naissance sont normales avec un poids et une taille moyennes respectivement de 3219 g et 51.1 cm chez les filles et 3182 g et 51.0 cm chez les garçons soulignant une croissance prénatale normale (Ranke, Heidemann et al. 1988). Cependant, 10% des enfants naissent avec une petite taille de naissance (Small for gestationnal age, SGA) (Ranke, Heidemann et al. 1988). Les oedèmes, parfois présents à la
naissance, pourraient masquer des anomalies du poids de naissance (Allanson 1987; Sharland, Burch et al. 1992).
Des difficultés alimentaires associées à une mauvaise prise pondérale sont présentes chez 75% des nourrissons ; ces difficultés peuvent être légères (15%), modérées (40%) ou sévères justifiant une nutrition entérale (20%) et peuvent persister jusqu’à l’âge de 18 mois.
L’évolution postnatale de la croissance staturale est identique dans les deux sexes avec une taille moyenne qui suit le troisième percentile jusqu’à 10 ans chez les filles et 12 ans chez les garçons. L’âge osseux est souvent retardé par rapport à l’âge chronologique d’environ deux ans et s’accompagne d’un retard pubertaire responsable d’un ralentissement de la vitesse de croissance dans les 2 sexes. Le pic de croissance pubertaire apparaît avec un retard de deux ans et est souvent réduit, la taille finale est atteinte à la fin de la deuxième décade.
La taille adulte spontanée est proche des -2 déviations standards (DS) (soit le 3ème percentile) de la population normale : 152.7 cm chez les femmes (n=55) et 162.5 cm chez les hommes (n=89) dans l’étude de Ranke et al. (Ranke, Heidemann et al. 1988) et 152.7 cm chez les femmes (n=25) et 167.4 cm chez les hommes (n=18) dans une étude plus récente (Shaw, Kalidas et al. 2007). A l’âge adulte, 30% des hommes et femmes ont une taille supérieure au 10ème percentile alors qu’environ 40% des hommes et 50% des femmes ont une taille en dessous du 3ème percentile (Noonan, Raaijmakers et al. 2003). L’existence ou la sévérité d’une cardiopathie associée ne semble pas avoir un impact majeur sur les caractéristiques de la croissance (Ranke, Heidemann et al. 1988).
Concernant la fonction génitale, l’âge moyen de la ménarche est de 14.6 ans, cependant la fertilité ne semble pas atteinte chez les femmes présentant un SN (Sharland, Burch et al. 1992). Chez le garçon, des anomalies de migration testiculaire sont fréquentes (75%) (Sharland, Burch et al. 1992) et des taux élevés de LH et FSH sont parfois présents en période prépubertaire (Theintz and Savage 1982). Chez l’adulte, des taux de FSL élevés et une faible qualité du liquide séminal ont été retrouvés, suggérant une anomalie de la spermatogenèse chez les garçons avec des testicules non descendus (Elsawi, Pryor et al. 1994).
1.2.2. Données hormonales
L’hormone de croissance (Growth Hormone, GH), sécrétée par l’antéhypophyse dans la circulation générale, se lie à des récepteurs membranaires au niveau des tissus cibles pour
stimuler la croissance somatique; la plupart de ses effets sont médiés par l’Insulin-Like Growth Factor (IGF) -I. L’IGF-I est transportée et stockée au niveau sanguin par des protéines porteuses dont l’IGF binding protein 3 (IGFBP-3) et l’acid labil subunit (ALS). Dans le SN, il n’existe habituellement pas de déficit en hormone de croissance (Growth hormone deficiency, GHD) mais des anomalies de l’axe somatotrope GH/IGF-I ont été observées.
Après stimulation pharmacologique, la sécrétion de GH est le plus souvent normale (Elders and Char 1976; Ahmed, Foot et al. 1991; Thomas and Stanhope 1993; Romano, Blethen et al. 1996; Kirk, Betts et al. 2001; Noordam, van der Burgt et al. 2001) ; certaines études retrouvent cependant des tests déficitaires chez environ un tiers des enfants (Otten BJ. ; Cotterill, McKenna et al. 1996; Romano, Blethen et al. 1996). L’étude du profil sécrétoire nocturne de la GH est également le plus souvent normale, cependant, certains enfants présentent des anomalies de la pulsatilité avec des pics irréguliers de GH et des dépressions moins profondes entre les pics (Ahmed, Foot et al. 1991; Tanaka, Sato et al. 1992; Noordam, van der Burgt et al. 2002).
Les taux d’IGF-I sont le plus souvent bas ou dans la normale basse (Ahmed, Foot et al. 1991; Tanaka, Sato et al. 1992; Noordam, van der Burgt et al. 2002), l’IGFBP-3 est normale (Cotterill, McKenna et al. 1996; Noordam, van der Burgt et al. 2001; Binder, Neuer et al. 2005; Limal, Parfait et al. 2006) et l’ALS est basse (Limal, Parfait et al. 2006).
1.2.3. Réponse au traitement par GH recombinante
Comme dans le syndrome de Turner et le retard statural secondaire à un SGA (Kamp, Kuilboer et al. 1993; Boguszewski, Rosberg et al. 1995), une diminution de la sensibilité à GH a été évoquée, c’est pourquoi, lors des protocoles thérapeutiques, des doses de GH humaine recombinante (human recombinant, rh) plus élevées que dans les déficits ont été proposées (0,05 à 0,066 versus 0,033 mg/kg/j dans les GHD).
Les premières études concernant le traitement par rhGH des enfants présentant un SN concernaient un petit nombre de patients (Cianfarani, Spadoni et al. 1987; Ahmed, Foot et al. 1991; Thomas and Stanhope 1993; Municchi, Pasquino et al. 1995) ou un traitement de courte durée (Cotterill, McKenna et al. 1996; De Schepper, Otten et al. 1997). Par la suite, plusieurs études concernant un nombre de patients plus important et un traitement prolongé d’au moins 3 ans ont été réalisées avec des résultats encourageants (Otten BJ. ; Romano, Blethen et al.
1996; Kirk, Betts et al. 2001; MacFarlane, Brown et al. 2001; Noordam, Van der Burgt et al. 2001; Limal, Parfait et al. 2006). Ainsi, le gain de taille moyen après 3 années de traitement est de presque 1 DS (0.8 à 0.9 DS). Il existe comme dans d’autres indications un épuisement du rattrapage initial avec une stabilisation (Cho, Thorvaldsen et al. 2001; Um, Frigerio et al. 2004) de la vitesse de croissance après la troisième année.
Sous traitement par rhGH, il existe une augmentation significative des taux d’IGF1 (de –1,6 à 0,3 DS après un an traitement dans l’étude de Limal et al.) (Ahmed, Foot et al. 1991; Limal, Parfait et al. 2006). La réponse au traitement est dose dépendante et ne semble pas liée aux données de la sécrétion GH avant traitement (Noordam, van der Burgt et al. 2001).
Deux études ont permis de préciser les tailles adultes après traitement par rhGH (Osio, Dahlgren et al. 2005; Noordam, Peer et al. 2008). Dans l’étude d’Osio et al. (n=18), la taille adulte est de –1.6 DS chez les filles et -0.9 DS chez les garçons soit un gain de taille respectivement de 1.8 et 1.5 DS ; 60% des enfants atteignent leur taille cible. Cet accroissement de taille est important au cours des premières années de traitement, sans effet de la dose de GH (0.033 vs 0.066 mg/kg/j). Après 3 ans de traitement, il n’existe plus de rattrapage mais le gain de taille obtenu est maintenu jusqu’au démarrage pubertaire. Du début de la puberté à la taille finale, une augmentation de la taille est obtenue chez les garçons du fait d’un pic de croissance pubertaire prolongé. Dans l’étude de Noordam et al. (n=28), la taille adulte moyenne est de -1.5 DS soit un gain de taille de 1.3 DS (9.5 cm chez les garçons et 9 cm chez les filles) ; 76% des patients ont une taille adulte supérieure à -2DS. L’accroissement de taille obtenu lors des 4 premières années de traitement est maintenu jusqu’à la taille adulte. La réponse au traitement est très variable, les facteurs prédictifs de bonne réponse sont l’âge de démarrage de la puberté et le nombre d’années de traitement avant le démarrage pubertaire.
Ces résultats sont semblables à l’accroissement de taille sous traitement par rhGH chez les filles avec un syndrome de Turner et les enfants petits nés SGA (Van Pareren, Mulder et al. 2003; van Pareren, de Muinck Keizer-Schrama et al. 2003).
La tolérance du traitement au niveau métabolique, hématologique et cardiaque à court et long terme est globalement bonne (Cotterill, McKenna et al. 1996; MacFarlane, Brown et al. 2001; Osio, Dahlgren et al. 2005; Noordam, Peer et al. 2008). Une augmentation modérée et transitoire de l’insulinémie à jeun sans modification des autres paramètres du métabolisme glucidique a été notée, de plus un enfant a développé un lymphome après 3 ans de traitement
(Osio, Dahlgren et al. 2005). Les inquiétudes concernant l’avance de maturation osseuse observée chez les patients traités plus importante que chez les contrôles n’ont pas été confirmées (Noordam, Van der Burgt et al. 2001; Noordam, Peer et al. 2008).
1.2.4. Hypothèses physiopathologiques du retard statural
Les anomalies de l’axe somatotrope observées et la réponse au traitement par rhGH ont fait évoquer plusieurs hypothèses :
a) Résistance partielle à GH par anomalie de signalisation post récepteur
L’association de taux normaux de GH, spontanés ou après stimulation, à des taux d’IGF-I bas suggère une insensibilité à GH (Growth hormone insentivity, GHI). Cette résistance est partielle comme le prouve la réponse au traitement par rhGH et pourrait être spécifique de l’IGF-I et l’ALS sans affecter l’IGFBP-3. Comme nous le verrons dans le chapitre suivant, l’identification dans le SN de mutations du gène PTPN11, codant pour une tyrosine phosphatase Shp2 impliquée dans la régulation du signal en aval du récepteur à GH (GH receptor, GHR), est en faveur de cette hypothèse.
b) Une anomalie hypothalamo-hypophysaire à type de dysfonction neurosécrétoire ne peut être exclue. En effet, les taux normaux d’IGFBP-3 (comme chez 70% des patients présentant un hypopituitarisme) et l’élévation des taux l’IGF-I sous traitement par rhGH sont compatibles avec cette hypothèse.
c) Enfin, une anomalie localisée au niveau de la plaque de croissance est également possible.
1.3. Cardiopathie
La cardiopathie congénitale est présente chez environ 80% des patients SN (Sharland, Burch et al. 1992; Marino, Digilio et al. 1999; Bertola, Kim et al. 2000). Le plus souvent, il s’agit d’une sténose des valves pulmonaires avec feuillets dysplasiques (60%) qui peut être isolée ou associée à d’autres anomalies cardiaques (Pernot, Marcon et al. 1987; Lin 1988). Une cardiomyopathie hypertrophique (hypertophic cardiomyopathy, HCM) peut également se développer (20%) tout au long de l’enfance. Plus rarement, sont retrouvées une communication inter auriculaire (CIA) (6 à 10%), une communication inter ventriculaire
(CIV) (5%), ou la persistance d’un canal artériel (3%) (Allanson 1987; Sharland, Burch et al. 1992). Enfin, certains patients présentent des anomalies du canal atrio-ventriculaire associées à une obstruction subaortique et des anomalies structurales de la valve mitrale (Marino, Digilio et al. 1999). L’électrocardiogramme (ECG) des patients SN retrouve des complexes QRS larges avec une négativation dans les dérivations précordiales gauches (60%), une déviation gauche de l’axe et des ondes Q géantes (Sanchez-Cascos 1983; Pernot, Marcon et al. 1987).
1.4. Anomalies hématologiques et de l’hémostase
A la naissance, une hépatomégalie et/ou une splénomégalie, non expliquées par une atteinte cardiaque, sont retrouvées chez 20 à 30% des enfants SN (Sharland, Burch et al. 1992) ; la fréquence de ces anomalies est de 50% lorsqu’une échographie abdominale est réalisée de manière systématique (George, Patton et al. 1993). Cette hépatosplénomégalie diminue souvent avec l’âge et pourrait être la manifestation d’un syndrome myéloprolifératif à minima.
Chez approximativement 1% des patients SN, un réel syndrome myéloprolifératif (leucémie myélomonocytaire chronique) est diagnostiqué devant des anomalies hématologiques modérées (thrombocytopénie et splénomégalie dans les premiers mois de vie) (Bader-Meunier, Tchernia et al. 1997; Fukuda, Horibe et al. 1997; Side and Shannon 1997; Choong, Freedman et al. 1999; Silvio, Carlo et al. 2002). Le plus souvent, ce syndrome myéloprolifératif régresse spontanément sans traitement, cependant, il peut parfois évoluer en
leucémie myélomonocytaire juvénile (LMMJ).
La LMMJ est une hémopathie rare (2% des hémopathies de l’enfant) mais agressive (6% de survie à 10 ans en l’absence de traitement) qui apparaît dans la petite enfance, avec une médiane au moment du diagnostic de 2 ans (Emanuel 2004). La LMMJ a une prévalence augmentée dans le SN bien qu’elle soit présente dans un faible pourcentage de cas (<1%) (Bader-Meunier, Tchernia et al. 1997; Fukuda, Horibe et al. 1997; Choong, Freedman et al. 1999). De plus, lorsqu’elle survient, l’évolution est souvent plus bénigne comparée aux LMMJ non associées au SN. Le rôle prédisposant du SN dans la survenue d’autres hémopathies, notamment les leucémies aigues lymphoblastiques (LAL), a également été
évoqué mais reste plus difficile à prouver compte tenu de la relative fréquence de ces tumeurs chez l’enfant (Piombo, Rosanda et al. 1993; Attard-Montalto, Kingston et al. 1994).
Enfin, des anomalies de l’hémostase sont souvent observées (20%). Ainsi, des
ecchymoses spontanées ou des saignements sont fréquents, notamment dans l’enfance ; cependant, des hémorragies sévères sont rares (3%). L’exploration de la coagulation peut retrouver un temps de saignement prolongé, des déficits des facteurs VIII, XI et XII, une thrombocytopénie et des anomalies fonctionnelles des plaquettes. Ces anomalies peuvent apparaître isolément ou en association (Sharland, Burch et al. 1992; Massarano, Wood et al. 1996). Chez les patients SN, il n’existe pas de corrélation entre les ecchymoses spontanées et les tests de coagulation. Ces tests ne peuvent donc être utilisés pour prédire le risque de saignement.
1.5. Anomalies du développement psychomoteur
Les nourrissons SN présentent souvent une hypotonie musculaire et une hyperlaxité articulaire responsables d’un retard moteur modéré : la tenue assise est acquise en moyenne à 10 mois, la marche à 21 mois et la parole à 31 mois (Sharland, Burch et al. 1992). La plupart des enfants (90%) suivent une scolarité normale mais certains présentent des retards des apprentissages (25%) et/ou nécessitent une pris en charge éducative spécialisée (10-15%). Un
retard mental modéré est présent chez 15 à 35% des patients: le quotient intellectuel est en
moyenne de 85 avec des variations importantes entre les patients (van der Burgt, Thoonen et al. 1999; Chan, Leedy et al. 2005). Ces enfants présentent des troubles visio-constructifs et une diminution des performances verbales. Des anomalies du comportement (déficit de l’attention, irritabilité, opposition) sont fréquemment observées.
1.6. Autres signes
Les autres signes observés chez les patients SN sont précisés dans le tableau 2. Les fréquences des différentes atteintes évaluées dans des cohortes anciennes, seront reprécisées dans le chapitre 3 à la lumière de la découverte des gènes impliqués dans cette pathologie.
Retard psychomoteur (25%), retard de langage (20%) et des apprentissages (25%), retard mental modéré (15-35%), troubles du comportement (hyperactivité, déficit de l’attention)
Neurologique
naevi (25%), taches café au lait (10%), lentigines (3%), ulérythème ophryogenes (14%), cheveux fins (30%)et épars (10%), foetal pads (67%)
Phanères
Mauvaise prise pondérale dans la petite enfance (75%)
Retard statural (80%) Croissance
Anomalies lymphatiques (lymphoedème, lymphangiectasie) (20%)
Lymphatique
Malformations tractus urinaire (sténose pyélo-urétérale et/ou hydronéphrose) (10%), cryptorchidie (70%)
Urogénitale
Pectus excavatus et/ou carinatus (70%), cubitus valgus (50%), hypermobilité articulaire (50%), clinobrachydactylie (30%), scoliose (15%), pieds varus equin (12%), synostose radioulnaire (2%)
Squelettique
Strabisme (60%), myopie et hypermétropie (60-70%), amblyopie (30%), nystagmus (10%), anomalies du segment antérieur (63%) et du fond d’œil (20%) Surdité (15-40%) de perception (otites) ou de transmission
Neurosensorielle
Hépatosplénomégalie (20-30%), syndrome myéloprolifératif (1%), LMMJ (<1%) Anomalies de la coagulation (20%)
Hématologique
Cardiopathie congénitale (80%): sténose pulmonaire (60%), HCM (20%), CIA (6-10%), CIV (5%), canal artériel persistant (3%), anomalie du canal atrioventriculaire, tétralogie de Fallot, anomalie mitrale
Anomalies ECG (60%)
Cardiaque
Retard psychomoteur (25%), retard de langage (20%) et des apprentissages (25%), retard mental modéré (15-35%), troubles du comportement (hyperactivité, déficit de l’attention)
Neurologique
naevi (25%), taches café au lait (10%), lentigines (3%), ulérythème ophryogenes (14%), cheveux fins (30%)et épars (10%), foetal pads (67%)
Phanères
Mauvaise prise pondérale dans la petite enfance (75%)
Retard statural (80%) Croissance
Anomalies lymphatiques (lymphoedème, lymphangiectasie) (20%)
Lymphatique
Malformations tractus urinaire (sténose pyélo-urétérale et/ou hydronéphrose) (10%), cryptorchidie (70%)
Urogénitale
Pectus excavatus et/ou carinatus (70%), cubitus valgus (50%), hypermobilité articulaire (50%), clinobrachydactylie (30%), scoliose (15%), pieds varus equin (12%), synostose radioulnaire (2%)
Squelettique
Strabisme (60%), myopie et hypermétropie (60-70%), amblyopie (30%), nystagmus (10%), anomalies du segment antérieur (63%) et du fond d’œil (20%) Surdité (15-40%) de perception (otites) ou de transmission
Neurosensorielle
Hépatosplénomégalie (20-30%), syndrome myéloprolifératif (1%), LMMJ (<1%) Anomalies de la coagulation (20%)
Hématologique
Cardiopathie congénitale (80%): sténose pulmonaire (60%), HCM (20%), CIA (6-10%), CIV (5%), canal artériel persistant (3%), anomalie du canal atrioventriculaire, tétralogie de Fallot, anomalie mitrale
Anomalies ECG (60%)
Cardiaque
2. Le syndrome LEOPARD
Le SL est une maladie autosomique dominante dont le nom acronymique se réfère aux caractéristiques cliniques les plus fréquentes : multiple Lentigines, Electrocardiographic-conduction abnormalities, Ocular hypertelorism, Pulmonary stenosis, Abnormal genitalia, Retardation of growth, sensorineural Deafness (Gorlin, Anderson et al. 1971). Ce syndrome est phénotypiquement très proche du SN et s’en différencie principalement par l’existence d’une atteinte cutanée (lentigines multiples) (figure 2) et d’une surdité. La prévalence de ce syndrome n’est pas connue, plus de 200 cas ont été rapportés à travers le monde.
Figure 2. Caractéristiques cliniques du syndrome LEOPARD
Noter l’hypertélorisme et les lentigines multiples au niveau du thorax (Sarkozy, Conti et al. 2004).
2.1. Anomalies de la croissance et de la puberté
Les mensurations à la naissance des patients SL sont le plus souvent normales, cependant environ un tiers d’entre eux présentent une macrosomie (définie par un poids de naissance au-dessus du 97ème percentile) (Digilio, Sarkozy et al. 2006). Par la suite, ces patients présentent un retard de croissance postnatal avec un retard statural (taille en dessous du 3ème percentile) chez environ 25% d’entre eux (Gorlin, Anderson et al. 1971; Voron, Hatfield et al. 1976). A l’âge adulte, 85% des patients auraient une taille en dessous du 25ème percentile (Sarkozy, Digilio et al. 2008).
Il n’existe actuellement pas de données sur le statut hormonal de ces patients.
Concernant la fonction génitale, une cryptorchidie bilatérale est présente chez 50% des garçons, mais des hypospadias et des hypoplasies génitales sont également retrouvés. Des
retards pubertaires et des ovaires hypoplasiques ont été rapportés chez la fille. La plupart des cas familiaux sont d’origine maternelle suggérant une réduction de la fertilité masculine (Voron, Hatfield et al. 1976; Digilio, Sarkozy et al. 2006).
2.2. Cardiopathie
Des malformations cardiaques sont retrouvées chez environ 80% des patients (Sarkozy, Conti et al. 2004; Limongelli, Pacileo et al. 2007; Limongelli, Sarkozy et al. 2008). Contrairement au SN, l’HCM est l’anomalie la plus fréquente (80%) alors que la sténose des valves pulmonaires n’est présente que chez 20% des patients. Cette HCM, qui est en général asymétrique et implique le ventricule gauche, est souvent associée à une obstruction d’éjection du ventricule gauche (40%) ; elle peut être congénitale mais le plus souvent se manifeste au cours de la seconde enfance. Des arrêts cardiaques et des morts subites ont été rapportés chez certains patients SL porteurs de HCM (Somerville and Bonham-Carter 1972; Woywodt, Welzel et al. 1998; Limongelli, Sarkozy et al. 2008).
Parmi les autres anomalies cardiaques, sont retrouvés des prolapsus de la valve mitrale (40%) et, moins fréquemment, des anomalies septales (CIA, CIV, canal atrioventriculaire), un élargissement du ventricule gauche isolé, une fibroélastose endocardique, des anomalies des artères coronaires, un anévrysme apical et non compaction du ventricule gauche (Coppin and Temple 1997; Limongelli, Pacileo et al. 2007).
Des anomalies de l’ECG sont observées chez 75% des patients. Ces anomalies sont en rapport avec l’HCM mais il existe également des troubles de la conduction (20%)(Limongelli, Pacileo et al. 2007).
2.3. Autres anomalies
Chez quelques patients SL, quelques cas de complications hématologiques (LAL, LAM, neuroblastome) ont été rapportés (Voron, Hatfield et al. 1976; Merks, Caron et al. 2005; Ucar, Calyskan et al. 2006; Laux, Kratz et al. 2008). Un mélanome malin (Seishima, Mizutani et al. 2007) et un choristome (tumeur de la cornée) bilatéral ont également été décrits (Choi, Yoo et al. 2003).
Le développement psychomoteur de ces enfants est souvent normal, cependant, 30% d’entre eux présentent un retard mental très modéré. La surdité est présente chez 20% des patients.
Chez ces patients sont également décrites des anomalies thoraciques (75%) et des malformations des voies urinaires (Digilio, Sarkozy et al. 2006).
3. Les autres syndromes apparentés au syndrome de Noonan
3.1. Le syndrome Cardio-Facio-Cutané
Le syndrome CFC décrit en 1986 est une maladie sporadique phénotypiquement très proche du SN (Reynolds, Neri et al. 1986; Wieczorek, Majewski et al. 1997). Les caractéristiques cliniques associent une dysmorphie faciale caractéristique (avec front haut, rétraction bitemporale, hypoplasie supraorbitaire), des anomalies ectodermiques (avec eczéma, ichtyose, hyperkératose, cheveux et sourcils fins, épars et fragiles) (figure 3), une cardiopathie et un retard mental (Roberts, Allanson et al. 2006). Les anomalies neurologiques sont constantes à des degré divers incluant hypotonie, retard psycho-moteur, retard de langage et difficultés d’apprentissage (Yoon, Rosenberg et al. 2007). Des critères diagnostiques ont été proposés par certains auteurs (Kavamura, Peres et al. 2002).
Figure 3. Caractéristiques cliniques du syndrome Cardio-Facio-Cutané
3.2. Le syndrome de Costello
Le SC décrit en 1971 est une maladie sporadique avec un phénotype beaucoup plus sévère que le SN (Costello 1977; Hennekam 2003).
La dysmorphie faciale se distingue par des traits grossiers, des joues pleines, des lèvres et une langue épaisses, une hypertrophie gingivale et une voix rauque (figure 4) (Delrue, Arveiler et al. 2002). La peau est douce et plissée au niveau du dos des mains, par contre les plis palmoplantaires sont particulièrement profonds et marqués. L’apparition de papillomes (péri-oraux, péri-nasaux ou péri-anaux), qui peut survenir entre l’âge de 2 ans et la fin de l’adolescence, est un signe clinique distinctif.
Figure 4. Caracéristiques cliniques du syndrome de Costello
Noter les traits grossiers du visage, les plis palmoplantaires épais et profonds (Delrue, Arveiler et al. 2002)
Les cardiopathies, retrouvées dans 60% des cas, sont essentiellement des HCM. Des troubles du rythme sont également fréquents.
Le retard psychomoteur est constant, avec une marche acquise vers 4 ans, et le retard mental moyen à léger (QI de 47 à 85) prédominant sur le langage. La plupart des enfants ont un comportement chaleureux, sociable et gai. Il existe souvent une macrocrânie.
Enfin, ce syndrome est associé à un risque de tumeur solide élevé (10 à 15%). La tumeur la plus fréquente est le rhabdomyosarcome, le plus souvent avec une histologie embryonnaire. Des neuroblastomes, des ganglioneuroblastomes et des carcinomes cellulaires de la vessie sont retrouvés moins fréquemment. Certains protocoles de dépistage systématique de ces tumeurs ont été proposés (DeBaun 2002; Gripp, Lin et al. 2006).
3.3. La neurofibromatose de type 1 et le syndrome de Legius
La NF1, ou maladie de Von Recklinghausen, est une maladie autosomique dominante dont la prévalence est de 1 pour 3000. Elle est caractérisée par l’association d’anomalies de la pigmentation (taches café au lait, lentigines axillaires), de neurofibromes cutanés et plexiformes, de nodules de Lisch au niveau de l’iris, d’une dysplasie osseuse et de gliomes des voies optiques (Williams, Lucas et al. 2009). Les autres signes fréquemment observés sont le retard statural, la macrocéphalie et des difficultés d’apprentissage et de comportement. La gravité de cette maladie est liée au risque d’apparition de tumeurs, comme les neuroblastomes et les rhabdomyosarcomes chez l’enfant. Plus rarement des leucémies de type LMMJ peuvent apparaître.
Certains patients présentant une NF1 présentent une dysmorphie faciale évocatrice de SN ; cette association a été parfois appelée syndrome Noonan-NF1 (figure 5) (Carey 1998).
Figure 5. Caractéristiques cliniques du syndrome Noonan-NF1
Noter chez le même patient l’existence d’une dysmorphie évocatrice de SN et de taches café au lait.
Le syndrome de Legius, ou « NF1-like », est une maladie autosomique dominante qui partage de nombreuses caractéristiques avec la NF1. Ce syndrome associe des taches café au lait, des lentigines axillaires et une macrocéphalie mais, contrairement à la NF1, il n’existe pas de neurofibromes, de nodules de Lisch ou de tumeur du système nerveux central (Brems, Chmara et al. 2007; Pasmant, Sabbagh et al. 2009).
Conclusion
Le SN et les syndromes apparentés représentent un groupe de syndromes génétiques phénotypiquement très proches, parfois difficiles à différencier sur le plan clinique. Pendant longtemps, s’est posée la question de l’expression variable d’une même maladie génétique ou de maladies impliquant des gènes différents. Récemment, la découverte des différents gènes impliqués dans ces syndromes a permis de répondre à cette question.
Chapitre II
Génétique du syndrome de Noonan et des syndromes apparentés :
les anomalies de la voie de signalisation Ras/MAPK
Dans tous ces syndromes, des mutations germinales constitutives de gènes codant pour des molécules impliquées dans la voie Ras/Mitogen Activated Protein Kinase (MAPK) ont été décrites ces dernières années. Dans ce chapitre, nous décrirons dans un premier temps les différentes molécules impliquées dans la voie de signalisation Ras/MAPK. Puis, nous préciserons les anomalies génétiques retrouvées dans le syndrome de Noonan et les syndromes apparentés.
1. La voie Ras/MAPK
La voie de signalisation Ras/MAPK joue un rôle majeur dans la régulation du cycle cellulaire, la prolifération, la différenciation, la survie et la mort cellulaires (Malumbres and Barbacid 2003; Giehl 2005); toutes ces étapes sont critiques pour permettre un développement normal. Cette voie transduit les signaux extracellulaires, véhiculés sous la forme de facteurs de croissance, d’hormones ou de petites molécules, à l’environnement intracellulaire. Elle a été particulièrement étudiée dans le contexte de l’oncogenèse puisque sa dérégulation est une des premières causes de cancers.
1.1. Généralités
Dans les cellules de mammifères, il existe 4 principales voies de signalisation MAPK qui ont été bien caractérisées (Johnson and Lapadat 2002). Ces cascades de signalisation sont constituées de 3 protéines kinases qui sont activées séquentiellement, en partie par des phosphorylations : une MAPK kinase kinase (MAPKKK), une MAPK kinase (MAPKK) et une MAPK (figure 6). La voie de signalisation extracellular signal-regulated kinase (Erk)/MAPK est activée par des facteurs de croissance et est particulièrement importante dans l’oncogenèse humaine (figure 7) alors que les voies de signalisation c-Jun N-terminal kinase (JNK), p38 et Erk5 sont activées en réponse à des signaux de stress provenant de l’environnement, notamment le choc osmotique ou les radiations ionisantes.
Figure 6. Les principales voies de signalisation MAPK (Roberts and Der 2007)
Figure 7. Activation de la voie Ras/MAPK
La liaison d’un ligand (facteur de croissance) à son récepteur (récepteur tyrosine kinase par exemple) entraîne la dimérisation et l’autophosphorylation du récepteur au niveau des résidus tyrosines du domaine cytoplasmique. Ces tyrosines phosphorylées servent alors de site d’ancrage pour des protéines possédant des domaines
phosphotyrosine binding (PTB) ou Src homology-2 (SH2) telle que la molécule adaptatrice Grb2. Sos1, qui est
associée de façon constitutive à Grb2, est alors recrutée au niveau de la membrane plasmatique à proximité de son substrat Ras. En facilitant le remplacement du GDP par le GTP, Sos1 stimule la formation de la forme active de Ras liée au GTP. Ras activé active alors Raf qui active à son tour la protéine kinase MAPK/Erk kinase (Mek) 1/2 qui active alors Erk 1/2. La protéine Erk 1/2 activée transloque au niveau du noyau où elle phosphoryle et régule divers facteurs de transcription, conduisant à des changements de l’expression des gènes cibles.
1.2. Description des molécules impliquées dans la voie Ras/MAPK
1.2.1. Ras et ses régulateurs
Les protéines Ras sont les membres fondateurs de la superfamille Ras des petites protéines G monomériques à activité guanosine triphosphate hydrolase (GTPase). Cette superfamille est constituée de 150 membres chez l’homme répartis dans 5 branches principales en fonction de leur similarité de séquence et de fonction : Ras, Rho, Rab, Ran et Arf (Wennerberg, Rossman et al. 2005). Les protéines Ras sont des modulateurs sophistiqués de processus cellulaires divers et complexes. Des mutations de RAS (Rat sarcoma) sont retrouvées dans de nombreuses tumeurs chez l’homme (Malumbres and Barbacid 2003). Un autre aspect important de ces protéines est leur implication dans les processus neuronaux comme les apprentissages, la mémoire et la plasticité synaptique (Atkins, Selcher et al. 1998; Selcher, Atkins et al. 1999).
1.2.1.1. Structure et fonction de Ras
Il existe 3 principales isoformes de Ras qui partagent 85% de leur séquence d’acides aminés : Harvey-Ras (HRas), Kirsten-Ras (KRas) et Neuroblastoma-Ras (NRas). Ces 3 isoformes sont exprimées de manière ubiquitaire, KRas étant exprimé pratiquement dans tous les tissus. Des modèles murins ont montré que HRas et NRas n’interviennent pas dans le développement alors que KRas est essentiel (Johnson, Greenbaum et al. 1997).
Ces différentes isoformes présentent 2 régions différentes : une région conservée en N-terminal (résidus 5-166) qui constitue le domaine catalytique, et une région hypervariable en C-terminal (résidus 167-188/189) qui constitue la séquence d’adressage membranaire (figure
8) (Wennerberg, Rossman et al. 2005).
Le domaine catalytique contient différentes régions fonctionnellement importantes permettant la liaison aux groupements phosphates des nucléotides, la liaison à la guanine des nucléotides, la liaison avec les effecteurs (boucle « switch I ») et la liaison aux activateurs de Ras (boucle « switch II »). La séquence de cette région est partagée par les membres de la superfamille Ras mais aussi les protéines Gsα et d’autres GTPases.
La région hypervariable contient un motif CAAX (C correspondant à une cystéine et A à une alanine) qui va permettre la modification post transcriptionnelle des protéines Ras et leur
adressage membranaire. En effet, le motif CAAX est la séquence de reconnaissance d’enzymes (farnesyltransferase et geranylgeranyltransferase I) qui catalysent l’addition covalente de lipides (farnesyl et d’isoprenoid geranylgeranyl respectivement) au niveau du résidu cystéine de ce motif.
Figure 8. Structure de la petite protéine G Ras.
Les domaines fonctionnels de la GTPase sont figurés sur le shéma (Wennerberg, Rossman et al. 2005)
Les protéines Ras fonctionnent comme des « switchs » moléculaires régulés par leur liaison à la guanosine diphosphate (GDP) ou à la guanosine triphosphate (GTP) (Mitin, Rossman et al. 2005). Ainsi, elles alternent entre une forme inactive liée au GDP et une forme active lié au GTP, cette dernière permettant la transduction du signal en interagissant avec des protéines situées en aval.
Les petites GTPases présentent une haute affinité de liaison pour le GDP et le GTP, mais possèdent de faibles activités intrinsèques pour l’échange GDP/GTP et l’hydrolyse du GTP. Le cycle GDP/GTP est donc contrôlé par 2 types de protéines régulatrices : les facteurs d’échange de nucléotides à guanine (Guanosine nucleotide exchange factors, GEFs), qui facilitent le remplacement du GDP par le GTP et stimulent la formation de la forme active de Ras liée au GTP, alors que les protéines activatrices de GTPases (GTPase activating proteins,
GAPs) stimulent la formation de la forme inactive liée au GDP. Les 2 états de liaison aux
nucléotides (Ras-GDP ou Ras-GTP) entraînent des changements conformationnels au niveau des régions switch I et II. C’est principalement à travers ces changements conformationnels de Ras que les protéines régulatrices et les effecteurs « reconnaissent » le statut nucléotidique de la GTPase (Mitin, Rossman et al. 2005).
1.2.1.2. Activation de Ras par la GEF Sos1
Les protéines Ras sont situées en aval de nombreux récepteurs transmembranaires: récepteurs à activité tyrosine kinase (receptor tyrosine kinase, RTKs), récepteurs aux cytokines, intégrines, récepteurs couplés aux protéines G hétérotrimériques (RCPG) et canaux calciques. L’activation de Ras par les RTKs, notamment le récepteur à l’EGF (EGF receptor, EGFR), est une des plus importante. Cette activation fait intervenir le module Grb2/Sos. L’activation en aval du RTK survient lors de la liaison d’un ligand (facteur de croissance) entraînant la dimérisation et l’autophosphorylation du récepteur au niveau des résidus tyrosines du domaine cytoplasmique. Ces tyrosines phosphorylées servent alors de site d’ancrage pour des protéines possédant des domaines phosphotyrosine binding (PTB) ou Src homology-2 (SH2) telle que la molécule adaptatrice Grb2. Grb2 est associée de façon constitutive à la GEF son of sevenless-1 (Sos1) qui est alors recrutée au niveau de la membrane plasmatique à proximité de son substrat Ras (Buday and Downward 1993).
Sos1 est une protéine complexe dans laquelle le domaine catalytique Cdc25 est précédé par les domaines histone folds (HF), Dbl homology-pleckstrin homology (DP-HP) et Ras exhange motif (REM), et suivis par la région polyproline (PxxP) qui lie des domaines SH3 (figure 9).
Figure 9. Structure et activation de Sos1 (Tartaglia, Pennacchio et al. 2007)
De manière basale, Sos1 est dans une conformation auto-inhibée faisant intervenir des interactions intra- et inter-moléculaires complexes impliquant notamment l’unité DH-PH (Sondermann, Soisson et al. 2004). Le mécanisme précis de l’activation de Sos1 est incomplètement compris. Sos se lie à la fois à Ras-GTP et Ras-GDP et son domaine Cdc25 déplace les nucléotides à guanine par un mécanisme biochimique complexe qui implique la formation de complexes binaires et ternaires avec Ras (Margarit, Sondermann et al. 2003). Le remplacement du GDP par le GTP, stimule la formation de la forme active de Ras liée au GTP.
1.2.1.3 Régulation négative de Ras par les GAPs p120RasGAP et Neurofibromine
Les deux GAPs de Ras les mieux caractérisées sont la p120RasGAP et la Neurofibromine (Nf) (Bernards and Settleman 2004). Ces deux molécules, co-exprimées de manière ubiquitaire, semblent jouer des rôles différents dans l’inactivation de Ras. La p120RasGAP possède deux domaines SH2 qui lui permette d’être recrutée au niveau de récepteurs activés ou de protéines adaptatrices. Au contraire, la Nf, qui ne possède pas de domaine SH2, présente une haute affinité pour de faibles niveaux de Ras lié au GTP. Ainsi, la Nf serait responsable du maintien de Ras sous sa forme inactive en l’absence de stimulation cellulaire alors que p120RasGAP régulerait de manière négative Ras en réponse à des facteurs de croissance.
1.2.2. La cascade de signalisation Raf/Mek/Erk
La forme active de Ras interagit avec de nombreuses molécules cibles (effecteurs), incluant notamment Raf, phosphatidylinositol 3-kinase (PI3K), RAL-guanine nucleotide dissociation stimulator (RAL-GDS) et la phospholipase Cε (PLCε). La voie de signalisation classique est la voie Ras/Raf/Mek/Erk impliquée dans la prolifération et la différenciation médiées par les facteurs de croissance.
1.2.2.1. Raf
Les sérine/thréonine kinases Raf sont les effecteurs directs de Ras et sont situées au sommet du module Raf/Mek/Erk. Il existe 3 isoformes de Raf (ARaf, BRaf et Raf-1 (ou CRaf)) qui présentent une structure semblable avec 3 régions conservées (conserved regions, CR) : CR1, CR2 et CR3 (figure 10).
CR1 contient un domaine de liaison à Ras (Ras binding domain, RBD) et un domaine riche en cystéine (cysteine-rich domaine, CRD) tous les deux impliqués dans le recrutement membranaire. CR2 est un domaine de régulation de phosphorylation, il contient un site de liaison (comprenant la sérine 259) pour la protéine 14-3-3 qui stabilise Raf dans sa forme inactive. Il a été montré que CR1 et CR2 constituent un domaine régulateur négatif ; ainsi, la suppression de ces 2 domaines conduit à l’activité oncogénique de Raf-1 (Heidecker, Huleihel et al. 1990). Le domaine CR3 contient le domaine catalytique avec le segment d’activation. Malgré ces caractéristiques structurales communes, les 3 isoformes de Raf diffèrent considérablement dans leur distribution tissulaire, leur mode d’activation et de régulation (Wellbrock, Karasarides et al. 2004). Ainsi, par exemple, alors que Raf-1 est exprimé de manière ubiquitaire, BRaf, qui possède l’activité kinase la plus forte, est surtout exprimé dans le cerveau. Les études d’invalidation des différentes isoformes de Raf chez la souris suggèrent des fonctions non redondantes au niveau du développement (Galabova-Kovacs, Kolbus et al. 2006).
A l’état basal, la portion N-terminale interagit et inactive le domaine kinase CR3 en C-terminal maintenant Raf dans une conformation auto-inhibée. Les dimères protéiques 14-3-3, qui lient les sérines 259 (au niveau du CR2) et 621 (en C-terminal) phosphorylées, stabilisent l’état auto-inhibé de Raf (Muslin, Tanner et al. 1996). L’activation de Raf par Ras induit la libération des dimères 14-3-3 et stimule dans le même temps la phosphorylation des sites d’activation du domaine kinase. L’effet cumulatif de ces phosphorylations, de la liaison à Ras et des interactions avec les lipides membranaires est de rompre l’auto-inhibition de Raf et de stabiliser la conformation active du domaine kinase (Dhillon, Meikle et al. 2002; Wellbrock, Karasarides et al. 2004). Des protéines phosphatases (PP) sont également impliquées dans l’activation de Raf. Ainsi, il a été montré que PP1 et PP2A régulent positivement l’activation de Raf en déphosphorylant le site de liaison de 14-3-3 au niveau de la sérine 259 permettant ainsi l’interaction avec Ras et le recrutement membranaire.
De manière intéressante, certaines études ont montré que l’hétérodimérisation avec BRaf contribue à l’activation de Raf-1 (Garnett, Rana et al. 2005). Cette hétérodimérisation avec BRaf induirait la conformation active de la kinase de Raf-1 en permettant la phosphorylation du segment d’activation de Raf-1. On ne sait pas actuellement si cette phosphorylation est causée par BRaf, une autre kinase ou une auto-phosphorylation de Raf-1. On ne sait également pas si Raf-1 peut moduler l’activité de BRaf et si l’hétérodimérisation concerne également ARaf.
1.2.2.2. Mek et Erk 1 et 2
Une fois activée, les protéines Raf sont capables de phosphoryler Mek 1/2 qui à son tour phosphoryle Erk 1/2. Contrairement, à la complexité de l’activation de Raf, Mek et Erk deviennent complètement actifs simplement par la phosphorylation du segment d’activation de leurs domaines kinases respectifs.
Mek 1 et 2 sont des kinases qui activent toutes les deux Erk 1 et 2 mais semblent ne pas avoir de fonction redondante. Les données génétiques de modèles murins indiquent que Mek 1 est essentiel pour le développement embryonnaire (Giroux, Tremblay et al. 1999) alors que Mek 2 ne l’est pas (Belanger, Roy et al. 2003). Les protéines Mek sont constituées d’un domaine régulateur en N-terminal et d’un domaine kinase (figure 11).
Figure 11. Structure de Mek 1 et 2 (Rodriguez-Viciana and Rauen 2008)
Erk 1 et 2 sont les derniers éléments de la cascade et exercent leur fonction sur de nombreuses molécules nucléaires et cytosoliques situées en aval. Les substrats de Erk 1 et 2 incluent des composants nucléaires, des facteurs de transcription, des protéines membranaires et des protéines kinases qui contrôlent la progression du cycle cellulaire, la différenciation et la prolifération (Yoon and Seger 2006).
1.3. Régulation de l’activation de la voie Ras/MAPK :
L’activation de la voie Ras/MAPK joue un rôle majeur dans des processus clés comme la différenciation et la prolifération, elle doit donc être précisément régulée pour permettre une réponse physiologique appropriée. Il existe divers mécanismes de contrôle permettant de moduler cette activation.
1.3.1. La tyrosine phosphatase Shp2
Shp2, codée par le gène PTPN11, est une protéine tyrosine phosphatase non récepteur exprimée de manière ubiquitaire et qui joue un rôle fondamental dans la régulation de voies de signalisation majeures (en particulier la voie Ras/MAPK) activées en réponse à de nombreux facteurs de croissance, cytokines et hormones. Le rôle de Shp2 dans la régulation positive de l’activation de la voie Ras/MAPK sera développé dans le chapitre 4.
1.3.2. Les protéines Sprouty et Spred
Chez l’homme, il existe 4 isoformes des protéines Sprouty (Spry 1 à 4) qui présentent deux domaines conservés : un domaine riche en cystéine en C-terminal permettant la localisation membranaire, et un domaine contenant un résidu tyrosine en N-terminal permettant, lorsqu’il est phosphorylé, l’interaction avec des molécules possédant un domaine SH2 (Kim and Bar-Sagi 2004). La divergence de séquence au niveau du domaine N-terminal entre les protéines Spry pourrait être à l’origine de leurs fonctions différentes en médiant des interactions protéines-protéines différentes. L’expression de ces protéines est induite par les mêmes voies de signalisation qu’elles contrôlent, constituant ainsi un rétrocontrôle négatif.
Des modèles d’invalidation génique ont montré que Spry exercerait une activité inhibitrice en aval des RTKs et en amont de Erk ; cependant, les mécanismes moléculaires précis sont incomplètement connus. Les protéines Spry pourraient interagir, par leur résidu tyrosine phosphorylé, avec la molécule adaptatrice Grb2. Ainsi, dans la signalisation induite par le fibroblast growth factor (FGF), l’association Spry/Grb2 entraîne une séquestration de Grb2 empêchant la transmission du signal (Hanafusa, Torii et al. 2002). Un autre mode d’action serait l’interaction des protéines Spry, par leur domaine riche en cystéine, avec le domaine catalytique de Raf. Cette interaction bloquerait l’activation de Raf par la protéine
kinase Cδ (PKCδ) mais pas l’activation de Raf dépendante de Ras (Sasaki, Taketomi et al. 2003).
Cependant, le rôle de régulateur négatif de Spry dans l’activation des RTKs reste controversé car leurs effets dépendent de l’isoforme de Spry, du récepteur activé, du type et du contexte cellulaire (Mason, Morrison et al. 2006). Ainsi, bien qu’étant considérés comme des régulateurs négatifs de l’activation de la voie Ras/MAPK en aval du FGFR, ces protéines réguleraient positivement cette voie en aval de l’EGFR (Waterman, Katz et al. 2002). Ce rôle positif pourrait s’expliquer par une inhibition de l’ubiquitination de l’EGFR (Wong, Fong et al. 2002).
Un domaine riche en cystéine partageant des similarités de séquence avec celui des protéines Spry (cysteine-rich domaine related to sprouty, SPR) a été identifié dans les protéines SPRY-related proteins with an EVH1 domain (Spred) dont il existe 2 isoformes (Spred 1 et 2). En plus du domaine SPR en C-terminal, ces protéines contiennent un domaine central de liaison à KIT (KIT-binding domain, KBD) et un domaine ENA/Vasodilatator-stimulated phosphoprotein (VASP) homology-1 (EVH1) en N-terminal (figure 12).
Figure 12. Structure des protéines Spred et Sprouty (Kim and Bar-Sagi 2004)
Le domaine SPR intervient dans la localisation membranaire des protéines Spred et dans l’interaction avec Raf. Le domaine EVH1 reconnaît et lie des séquences riches en proline, cependant les ligands spécifiques des protéines Spred ne sont pas encore identifiés. Enfin, la fonction physiologique du domaine KBD est encore inconnue.
Les protéines Spred semblent agir principalement en aval de Ras en inhibant la phosphorylation et l’activation de Raf (Wakioka, Sasaki et al. 2001). Ainsi, Spred s’associe à Ras ce qui n’empêche pas l’activation de Ras ni le recrutement membranaire de Raf. Par contre, Spred prolonge l’association de Raf à Ras ce qui pourrait diminuer son activation en altérant sa distribution cellulaire.
2. Les anomalies génétiques de la voie Ras/MAPK
Des mutations germinales constitutives de gènes codant pour des molécules impliquées dans la voie Ras/MAPK sont retrouvées dans le syndrome SN et les syndromes apparentés (figure 13).
Figure 13. Les anomalies constitutives des molécules impliquées dans la voie Ras/MAPK à l’origine des syndromes Neuro-Cardio-Facio-Cutanés
Ainsi, il a été proposé de regrouper l’ensemble de ces syndromes sous le nom de « syndromes Neuro-Cardio-Facio-Cutanés » (NCFC) (Bentires-Alj, Kontaridis et al. 2006). Cependant, la classification nosologique de ces syndromes continue à être discutée. Actuellement, il semble admis que le SN est causé par des mutations de 3 gènes (PTPN11, SOS1 et RAF1), le SL par des mutations de 2 gènes (PTPN11 et RAF1), le syndrome CFC par des mutations de 3 autres gènes (BRAF, MEK 1 et 2), et le syndrome de Costello par des mutations de HRAS. Les patients présentant des mutations de KRAS sont plus difficiles à classer du fait de phénotypes à la frontière entre les SN, CFC et Costello (Neri, Allanson et al. 2008). Cependant, dans la littérature, de nombreuses exceptions ont été décrites pour d’autres gènes : mutation de SOS1 chez un patient diagnostiqué CFC (Nystrom, Ekvall et al. 2008) ou mutation de BRAF chez des patients diagnostiqués SN (Razzaque, Nishizawa et al. 2007; Nystrom, Ekvall et al. 2008).